
Effective enhancement of the electrochemical performance of layered cathode Li1.5Mn0.75Ni0.25O2.5 via a novel facile molten salt method†
Zhuo Zhenga,
Wei-Bo Huaa,
Shi-Xuan Liaoa,
Yan-Jun Zhonga,
En-Hui Wanga,
Bin-Bin Xub,
Hua-Kun Liuc and
Ben-He Zhong*a
aCollege of Chemical Engineering, Sichuan University, No. 24 South Section 1, Yihuan Road, Chengdu, 610065, China. E-mail: zhongbenhe@163.com
bDepartment of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Fujian 361005, China
cInstitute for Superconducting and Electronic Materials, Australian Institute for Innovative Materials, University of Wollongong, Innovation Campus, North Wollongong, NSW, Australia
First published on 22nd June 2015
Abstract
A series of nanocrystalline lithium-rich cathode materials Li1.5Mn0.75Ni0.25O2.5 have been prepared by a novel synthetic process, which combines the co-precipitation method and a modified molten salt method. By using a moderate excess of 0.5LiNO3–0.5LiOH eutectic salts as molten media and reactants, the usage of deionized water or alcohol in the subsequent wash process is successfully reduced, compared with the traditional molten salt method. The materials with different excess Li salt content, Li/M (M = Ni + Mn) = 1.55, 1.65, 1.75, 1.85, 1.95, 2.05, molar ratio, show distinct differences in their structure and charge–discharge characteristics. The structural characterization demonstrates that the sample with a ratio of Li/M = 1.85 has a more well-defined α-NaFeO2 structure and a more enlarged Li layer spacing. It also exhibits the best comprehensive electrochemical behavior with the highest coulombic efficiency, the best rate capability and optimal cycling stability. More specifically, it delivers a dramatically improved initial coulombic efficiency of 87.86%, and a discharge capacity of 129 mA h g−1 even at an ultra-high current density of 2000 mA g−1 (10C). Meanwhile a superior cycling stability is also observed with a high discharge capacity of 251 mA h g−1 and a retention of 98% at 0.2C after 50 cycles. Our results reveal that this method is facile and feasible to synthesize a high rate and high capacity lithium-rich material.
1. Introduction
Rechargeable lithium ion batteries (LIBs) with high energy and high power densities are essential for the fast development of portable electronics, electric vehicles (EVs) and hybrid electric vehicles (HEVs).1–7 To meet the demand, the development of a new cathode with a high energy density is strongly desired. Currently, the family of lithium-rich layered oxides Li1+yM1−yO2 (also written as xLi2MnO3·(1 − x)LiMO2, M = Ni, Co, Mn) have attracted much attention, due to their encouraging high reversible capacity over 250 mA h g−1 when initially charged above 4.5 V.8–10 Among these lithium-rich oxides, the Li-rich Mn-based nickel oxide Li1.5Mn0.75Ni0.25O2.5 is the most appealing, due to its high discharge capacity, low cost, good safety and lower toxicity.11–13 However, it suffers several serious drawbacks which need to be resolved, such as its intrinsically inferior rate capability, poor cycle performance, a large irreversible capacity loss (ICL) in the initial cycle, and voltage decay during long-term cycling resulting from phase transformation from a layered to a spinel/layered intergrown structure.14–16To address these obstacles, many strategies have been developed, especially with regard to the synthetic methods. At present, many synthetic routes have been reported,17–20 including solid-state reaction, co-precipitation method, sol–gel method, molten salt synthetic method and others. Among these methods, the molten salt method is a simple and versatile way to prepare highly crystalline cathode materials by using molten salt as the solvent and/or reactants.21–25 Additionally, in the molten liquid environment, reactions are primarily decided by chemical equilibria and proceed much faster than diffusion-controlled solid-state reactions.20,25 Despite these advantages, some inherent shortcomings also exist. For example, the eutectic molten salts of KCl, NaCl, KNO3 or LiNO3–LiCl are required in large excess compared to the starting transition metal salts in order to provide sufficiently liquid conditions to promote transition metal atom diffusion; usually a molar ratio of n(starting materials)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(molten salt) is 1:3 or 1:4, or even higher is used.20–23 Hence, the use of molten salts would not only cause unnecessary water waste and increase environmental pressure, as much deionized water or alcohol is needed to wash the product to remove the excess Li-salts or KCl or NaCl salts, but it also increases the cost from an economic perspective as a large excess of molten salt is consumed. Furthermore, although the reactions in molten media are decided by chemical equilibria, if the starting materials cannot be mixed thoroughly then inhomogeneous phases appear in the product and deteriorate the electrochemical performance. Generally, a stoichiometric ratio of Li salt and transition metal (Mn, Ni, Co) salts and a large excess of the molten salt are mixed through simple mechanical grinding in the traditional molten salt method,22,24,25 therefore, it is very difficult to achieve molecule-level mixing of these starting materials. Fortunately, the co-precipitation method can compensate for this defect and easily realize molecule-level mixing. This method is considered to be the optimum way to obtain a homogeneous and integrated structure, which is the most crucial factor to achieve better electrochemical properties.26
:n(molten salt) is 1:3 or 1:4, or even higher is used.20–23 Hence, the use of molten salts would not only cause unnecessary water waste and increase environmental pressure, as much deionized water or alcohol is needed to wash the product to remove the excess Li-salts or KCl or NaCl salts, but it also increases the cost from an economic perspective as a large excess of molten salt is consumed. Furthermore, although the reactions in molten media are decided by chemical equilibria, if the starting materials cannot be mixed thoroughly then inhomogeneous phases appear in the product and deteriorate the electrochemical performance. Generally, a stoichiometric ratio of Li salt and transition metal (Mn, Ni, Co) salts and a large excess of the molten salt are mixed through simple mechanical grinding in the traditional molten salt method,22,24,25 therefore, it is very difficult to achieve molecule-level mixing of these starting materials. Fortunately, the co-precipitation method can compensate for this defect and easily realize molecule-level mixing. This method is considered to be the optimum way to obtain a homogeneous and integrated structure, which is the most crucial factor to achieve better electrochemical properties.26
In this paper we firstly propose a novel process that combines the hydroxide co-precipitation method and the molten salt method to prepare a Li1.5Mn0.75Ni0.25O2.5 electrode. It is worth emphasising that the molten salt method herein differs from the traditional one, mainly because we do not need a large excess of molten salt so that water consumption at the washing stage can be drastically reduced. This is because the transition metals have already achieved molecule-level mixing in the Mn0.75Ni0.25(OH)2 precursor using a co-precipitation method. As far as we know, there have been few reports which investigate the effect of the amount of salt flux on the final materials. Hence, we employ a series of moderate excesses of 0.5LiNO3–0.5LiOH as the eutectic molten media and reactants to synthesize the Li1.5Mn0.75Ni0.25O2.5 materials. The structure, morphology and electrochemical performance of these materials with gradually incremented amounts of Li salts are extensively investigated. Furthermore, we find that when the molar ratio of Li/M (M = Ni + Mn) is 1.85, the sample exhibits the best rate capability and optimal cycling stability with the lowest irreversible capacity loss (ICL) in the initial cycle.
2. Experimental section
2.1 Sample preparation
The layered Li1.5Mn0.75Ni0.25O2.5 material was prepared by a novel method that combined the hydroxide co-precipitation method and the molten salt method. Firstly, the Mn0.75Ni0.25(OH)2 precursor was prepared by a co-precipitation method. NiSO4·6H2O, MnSO4·H2O (Ni:Mn = 1:3, molar ratio) were dissolved in distilled water and then pumped into continuous stirred tank reactors (CSTR) under nitrogen atmosphere. Meanwhile, a NaOH solution (8 M) and a NH3·H2O solution (2 M) were separately fed into the CSTR at 50 °C. The pH value was carefully controlled at 11.0 ± 0.2 and the stirring speed was maintained at around 800 rpm. Afterwards, the co-precipitated particles were washed with hot distilled water several times to remove the residual Na+ and SO42−. The powders were dried in a vacuum oven at 90 °C for 12 h. Then, the 0.5LiNO3–0.5LiOH eutectic mixture, used as a molten medium and a lithium source, was mixed with the Mn0.75Ni0.25(OH)2 precursor. The molar ratios of the eutectic salts to the starting material (n(Li):n(M)) were set as 1.55, 1.65, 1.75, 1.85, 1.95, 2.05, respectively. All samples had 5% excess Li salt to compensate for possible Li loss during the calcinations. The obtained materials were designated as L0, L1, L2, L3, L4 and L5, respectively. The mixture was first heated at 500 °C for 6 h, and then calcined at 850 °C for 12 h in air. Afterwards, these materials were washed with a small amount of hot distilled water, and finally dried in a vacuum oven at 90 °C for 10 h to obtain the final lithiated compounds.
2.2 Sample characterization
The lithium content of these compounds was characterized by inductively coupled plasma-atomic emission spectrometry (ICP-AES, SPECTRO ARCOS FHS12). Powder X-ray diffraction (XRD, PW1730, Cu Kα radiation, λ = 1.5418 Å) was employed to identify the crystalline structures of the as-prepared compounds. The XRD data were collected in the range of 10–70° 2θ. The particle size and the morphological features were observed using scanning electron microscopy (SEM, S4800). The chemical valence state of Ni and Mn were determined using X-ray photoelectron spectroscopy (XPS, PHI QUANTUM2000) with a monochromatic Al-Kα anode source, and the binding energies (BE) were calibrated with reference to the C 1s spectrum of carbon (284.60 eV). The microstructures of these composites were observed using transmission electron microscopy (TEM, JEM-2100).2.3 Electrochemical measurement
The electrochemical properties of these cathode materials were measured with CR2032-type coin cells, which consist of an oxide cathode, a Li metal anode and a porous polypropylene separator (Celgard 2400) with a 1 M LiPF6 solution in EC–DMC (1:1 in volume rate) as the electrolyte. The cathodes were prepared by mixing the oxide powder, carbon black and polyvinylidene difluoride (PVDF) binder (80:13:7, weight ratio) in N-methyl pyrrolidone (NMP). The obtained slurry was casted onto aluminum foil and dried in a vacuum oven at 100 °C for 10 h. The foil was cut into discs with diameters of 14 mm and then pressed prior to use. The loading of the active material in the electrode was 3–4 mg cm−2. All coin cells were assembled and sealed in an argon-filled glove box. The cells were charged and discharged galvanostatically between 2.0 and 4.8 V (vs. Li/Li+) at room temperature with current densities of 0.1C, 0.2C, 0.5C, and 1C, respectively. Above discharge rates of 1C, the cells were charged at 1C and discharged at the corresponding rates from 3C to 10C (1C = 200 mA g−1). Electrochemical impedance spectroscopic (EIS) measurements were tested on an electrochemical workstation (Zennium, IM6) in the frequency range of 100 kHz to 10 mHz with an alternating current amplitude of 5 mV.
3. Results and discussion
ICP-AES analysis data for the atomic compositions of the as-synthesized materials are listed in Table 1. It illustrates that the ratios of Ni:Mn and Li/[Ni + Mn] in all the samples are approximately consistent with the expected Li1.5Mn0.75Ni0.25O2.5 stoichiometry within experimental errors.
:Mn and Li/[Mn + Ni] molar ratios in the samples
| Sample | Ni:Mn |
Li/[Ni + Mn] |
|---|---|---|
| L0 | 0.248:0.750 |
1.498 |
| L1 | 0.250:0.751 |
1.499 |
| L2 | 0.249:0.751 |
1.501 |
| L3 | 0.251:0.750 |
1.502 |
| L4 | 0.252:0.752 |
1.502 |
| L5 | 0.252:0.753 |
1.504 |
Fig. 1 shows the XRD patterns for the samples. As shown in Fig. 1a, all the samples exhibit similar XRD patterns. The majority of the diffraction peaks are relatively strong and can be well indexed to the rhombohedral layered phase (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) which is normally taken as the layered characteristic of the LiMO2 (M = Ni, Mn) structure,27,28 while a series of weak reflections at 20–23° (2θ value) are consistent with the hexagonal LiMn6 super-ordering in the Li2MnO3 monoclinic phase with C2/m symmetry.27,28 Combined with the ICP analysis above (Table 1), it can be concluded that the Li1.5Mn0.75Ni0.25O2.5 lithium-rich material has been successfully synthesized via this novel molten salt method. Compared with the traditional molten salt method, less molten salt is used in our synthetic route, but interestingly, highly crystalline materials can still be prepared at similar calcination temperatures to the traditional molten salt method. This can be ascribed to the molecule-level mixing of the transition metal salts which occurs during the co-precipitation process. The diffraction peaks of the Li2MnO3 phase change significantly (marked by yellow dotted lines in Fig. 1a), so a detailed comparison of the peak change of the selected patterns in the 2θ range 20.0–22.0° is shown in Fig. 1b. As can be seen, the peaks of the Li2MnO3 phase become clearer with increasing Li content, indicating a higher crystallinity. This can be legitimately attributed to the use of the 0.5LiNO3–0.5LiOH eutectic mixture with a lower melting point that can introduce some liquid to the system, thus promoting the reaction activation and lowering the reaction temperature.21,23,25 Therefore at the same calcination temperature, the more 0.5LiNO3–0.5LiOH eutectic mixture that is added, the higher the degree of crystallization achieved. Rietveld refinements within the layered LiMO2 (Rm) phase were conducted using TOPAS software to illustrate the variations from the view of the lattice parameters. The refinement results are listed in Table 2. The lattice constant a shows little change, however, the c-lattice parameter is obviously changed. The c-lattice parameter is perpendicular to the Li layer in the layered structure, therefore, its increase signifies an enlargement of the Li layer spacing.29 The L3 sample exhibits the largest c-lattice parameter, which may cause a substantially higher Li diffusivity.29 Generally, the c/a value is associated with the degree of trigonal distortion: a high c/a value is preferred for well-defined hexagonal α-NaFeO2 structures.30–32 Therefore, it can be inferred that the L3 sample has a better layered structure than the others.
m) which is normally taken as the layered characteristic of the LiMO2 (M = Ni, Mn) structure,27,28 while a series of weak reflections at 20–23° (2θ value) are consistent with the hexagonal LiMn6 super-ordering in the Li2MnO3 monoclinic phase with C2/m symmetry.27,28 Combined with the ICP analysis above (Table 1), it can be concluded that the Li1.5Mn0.75Ni0.25O2.5 lithium-rich material has been successfully synthesized via this novel molten salt method. Compared with the traditional molten salt method, less molten salt is used in our synthetic route, but interestingly, highly crystalline materials can still be prepared at similar calcination temperatures to the traditional molten salt method. This can be ascribed to the molecule-level mixing of the transition metal salts which occurs during the co-precipitation process. The diffraction peaks of the Li2MnO3 phase change significantly (marked by yellow dotted lines in Fig. 1a), so a detailed comparison of the peak change of the selected patterns in the 2θ range 20.0–22.0° is shown in Fig. 1b. As can be seen, the peaks of the Li2MnO3 phase become clearer with increasing Li content, indicating a higher crystallinity. This can be legitimately attributed to the use of the 0.5LiNO3–0.5LiOH eutectic mixture with a lower melting point that can introduce some liquid to the system, thus promoting the reaction activation and lowering the reaction temperature.21,23,25 Therefore at the same calcination temperature, the more 0.5LiNO3–0.5LiOH eutectic mixture that is added, the higher the degree of crystallization achieved. Rietveld refinements within the layered LiMO2 (Rm) phase were conducted using TOPAS software to illustrate the variations from the view of the lattice parameters. The refinement results are listed in Table 2. The lattice constant a shows little change, however, the c-lattice parameter is obviously changed. The c-lattice parameter is perpendicular to the Li layer in the layered structure, therefore, its increase signifies an enlargement of the Li layer spacing.29 The L3 sample exhibits the largest c-lattice parameter, which may cause a substantially higher Li diffusivity.29 Generally, the c/a value is associated with the degree of trigonal distortion: a high c/a value is preferred for well-defined hexagonal α-NaFeO2 structures.30–32 Therefore, it can be inferred that the L3 sample has a better layered structure than the others.
 | ||
| Fig. 1 (a) XRD patterns of the compounds; (b) magnified area in the 20.0–22.0° 2θ region. | ||
| Sample | a (Å) | c (Å) | c/a | Vol. (Å3) | Rwp (%) |
|---|---|---|---|---|---|
| L0 | 2.8550 | 14.2405 | 4.9879 | 100.52 | 4.10 |
| L1 | 2.8571 | 14.2580 | 4.9904 | 100.80 | 3.97 |
| L2 | 2.8555 | 14.2567 | 4.9927 | 100.65 | 3.85 |
| L3 | 2.8547 | 14.2609 | 4.9956 | 100.46 | 4.17 |
| L4 | 2.8580 | 14.2444 | 4.9840 | 100.76 | 4.28 |
| L5 | 2.8584 | 14.2377 | 4.9810 | 100.68 | 4.46 |
X-ray photoelectron spectroscopy (XPS) measurements were carried out to gain more insight into the oxidation states of the transition metals in the as-prepared samples. Fig. 2 shows the typical XPS spectra of Ni 2p and Mn 2p of the samples. All the XPS spectra were corrected using C 1s at 284.60 eV. For the L0 sample, the Ni 2p3/2 and Mn 2p3/2 peaks are observed at 854.5 eV and 642.2 eV, respectively, which are consistent with previous reports for Ni2+ and Mn4+ in similar oxide cathode materials.33–35 However, these peaks in other samples are shifted with the increased Li content. The Mn 2p3/2 peaks are slightly shifted toward lower binding energies, indicating that some lower oxidation states than Mn4+ exist. Correspondingly, a similar peak shift in the Ni 2p3/2 spectra toward higher binding energies is also observed with increasing Li content. The increasingly significant presence of Ni3+ and Mn3+ may be ascribed to the increase of 0.5LiNO3–0.5LiOH salts which would be decomposed to Li2O during the high temperature calcination; excess Li2O attaches on the surface of the reactant particles, preventing contact with oxygen, thus resulting in the incomplete oxidation of manganese. It is likely that the increase in the oxidation state of Ni is a compensation for the reduction in oxidation state of the Mn ions.
 | ||
| Fig. 2 XPS spectra of Ni 2p and Mn 2p for the samples. | ||
Fig. 3a shows the SEM images of the Ni0.25Mn0.75(OH)2 precursor. The micrographs show that the precursor consists of agglomerates at the micron level, which consist of hexagonal nanoplates with a thickness of about 40 nm, and lateral dimensions ranging from 200 to 500 nm. Fig. 3b shows the SEM images of the as-prepared samples. As the Li content increases, the particle sizes in terms of the thickness and the lateral dimensions gradually increase. Take the thickness of the nanoplates for example: the thickness distribution and the average thickness of the samples are shown in Fig. S1 in the ESI.† As can be seen, the average thicknesses of samples L0, L1 and L2 are similar; in contrast, a dramatic expansion in the thicknesses of samples L3, L4 and L5 is observed, especially the L5 sample, with an average thickness as high as 244 nm. The significant change in particle size can be ascribed to the excess Li salts that can lower the reaction temperature and promote the reaction activation, as mentioned in the XRD analysis. In general, a smaller particle size could indicate a better electrochemical performance.36 Nevertheless, the particle crystallinity also affects the stability of the parent structure during the repeated Li+ ion insertion/extraction processes, thus affecting the electrochemical properties of the material. Although samples L0, L1 and L2 have a smaller primary particle size than the L3 sample, their comparatively lower crystallinity is shown in Fig. 1, and may restrict their electrochemical behaviour to a certain degree. Moreover, the average thicknesses of samples L4 and L5 increases to 200–300 nm, which is much bigger than the L3 sample (157 nm), and a larger thickness indicates a much longer Li+ diffusion path which is related to a poorer rate capability. Fig. 3c shows the SAED images of samples L0, L3 and L5. It is clear that all the SAED patterns consist of two sets of reflections. The strong hexagonal reflections can be indexed in six-index notation (marked by the yellow circles), indicating an α-NaFeO2 layered structure (Rm).37 The weak reflections (marked by a red circle), which are associated with Li ordering in Li2MnO3-like domains,37 are clearly identified between the two bright spots of the layered trigonal symmetry. The SAED patterns also reveal that the samples prepared by this novel method are single crystalline.
 | ||
| Fig. 3 SEM images of the (a) Ni0.25Mn0.75(OH)2 precursor, (b) samples L0–L5; (c) SAED images of samples L0, L3 and L5. | ||
Fig. 4 shows the first charge–discharge curves during the electrochemical cycling of the as-prepared cathode materials at a rate of 0.1C (20 mA g−1) between 2.0 and 4.8 V. Clearly, the initial charge profiles of the samples show a smooth sloping region below 4.5 V and a high voltage plateau at around 4.5 V. The sloping region below 4.5 V can be ascribed to Li+ deintercalation from the layered structure by oxidation of Ni2+ to Ni4+.38,39 The plateau at around 4.5 V can be attributed to the Li2MnO3 activation which leads to a large initial irreversible capacity loss (ICL).38,39 The first coulombic efficiency increases from 77.13% in L0 to 87.86% in L3. However, for samples L4 and L5, the ICLs become larger, with lower coulombic efficiencies of 76.85% and 75.52%, respectively. The enhanced coulombic efficiency of the L3 sample can be reasonably attributed to the more well-defined α-NaFeO2 structure which can improve the lattice stability and alleviate the irreversible loss of Li2O during the Li2MnO3 activation.
 | ||
| Fig. 4 Initial charge–discharge curves of the cathodes when cycled between 2.0 and 4.8 V at 0.1C (20 mA g−1). | ||
Fig. 5a shows the continuous cycling results at incremental rates from 0.1C to 10C then recovering back to 0.2C. As can be seen, the L3 sample exhibits the best rate capability. It yields maximal discharge capacities of 261, 256, 238, 214, 187 and 154 mA h g−1 at 0.1C, 0.2C, 0.5C, 1C, 3C and 5C, respectively. More surprisingly, a maximal capacity of 129 mA h g−1 is still achieved at 10C (2000 mA g−1). This outstanding rate capability was also observed previously in excellent reports,10,40,41 which have similar nanoplate morphologies to our research. However, the L4 and L5 electrodes show poorer rate capability than the other samples. This result is consistent with the above XRD and SEM analyses (Fig. 1 and 3), and confirms that these two samples have an inferior layered structure and a large nanoplate size. In order to compare the rate capabilities more intuitively, the capacity retention of these samples at 0.2C, 0.5C, 1C, 3C, 5C and 10C rates relative to the 0.1C rate are shown in Fig. 5b (note: the capacity retention at 0.1C is defined as 100%, and the calculation equation can be written as Q(□C)/Q(0.1C), where Q represents the discharge capacity). It is also notable that the L3 sample delivers the highest capacity retention, with capacity retentions of 98%, 91%, 82%, 72%, 59% and 50% at 0.2C, 0.5C, 1C, 3C, 5C and 10C, respectively.
 | ||
| Fig. 5 (a) Rate capability of the samples; (b) capacity retention at different rates (note, retention at 0.1C is 100%). | ||
Fig. 6 shows the cycling performances of the samples at 0.2C between 2.0 and 4.8 V (inset), and the discharge curves for selected cycles. The L3 sample exhibits the optimal cycling stability, delivering a discharge capacity of nearly 256 mA h g−1 at first cycle, and maintaining 251 mA h g−1 after 50 cycles with a capacity retention of 98%. Similar to the rate performance, the L4, L5 samples display the poorest cycle capability, with capacity retentions of 86% and 83%, respectively. Additionally, in order to investigate exactly the voltage fading upon cycling, the 1st and 50th discharge profiles of these samples, after normalization of the capacity, are shown in Fig. S2.† Clearly, the discharge curves of the L3 sample exhibit the best consistency and this sample delivers the lowest voltage decay with Δ = 0.091 V for the voltage fading at the half capacity. This analysis indicates that the fast voltage fading upon cycling can be alleviated in the L3 sample, and that this novel molten salt method provides a simple and convenient way to address the huge challenge of layered-spinel intergrowth that could significantly lower the energy density of the whole battery.14,16
 | ||
| Fig. 6 Cycling stability of the samples at 0.2C between 2.0 and 4.8 V (inset), and the discharge curves for selected cycles. | ||
Electrochemical impedance spectroscopy (EIS) was performed to investigate the difference in electrochemical performance of the samples. The measurements were carried out after the electrochemical examination in Fig. 5a. All Nyquist plots are shown in Fig. 7a, and the corresponding equivalent circuit is presented in the inset. In this equivalent circuit, Rs represents the resistance of the electrolyte and cell components, Rct corresponds to the charge transfer resistance in the electrode–electrolyte interfaces, and W1 is the Warburg impedance which represents the Li+ ion diffusion process in the electrode materials.42–44 Each impedance spectrum is fitted well with the suggested equivalent circuit model, and the histogram (Fig. 7b) shows the variation of the corresponding fitted parameters. Similarly, by monitoring the samples, it was found that Rs remains almost unchanged, whereas Rct decreases from samples L0 to L3, and then increases quickly to sample L5. The much lower Rct value for the L3 sample is beneficial for reversible lithium ion deintercalation and intercalation during repeated charge and discharge processes, leading to a better electrochemical performance.42–44 The lower Rct for the L3 sample can be ascribed to the expansion of the c-parameter and the well-defined α-NaFeO2 layered structure leading to faster charge-transfer reactions in the electrode–electrolyte interfaces.
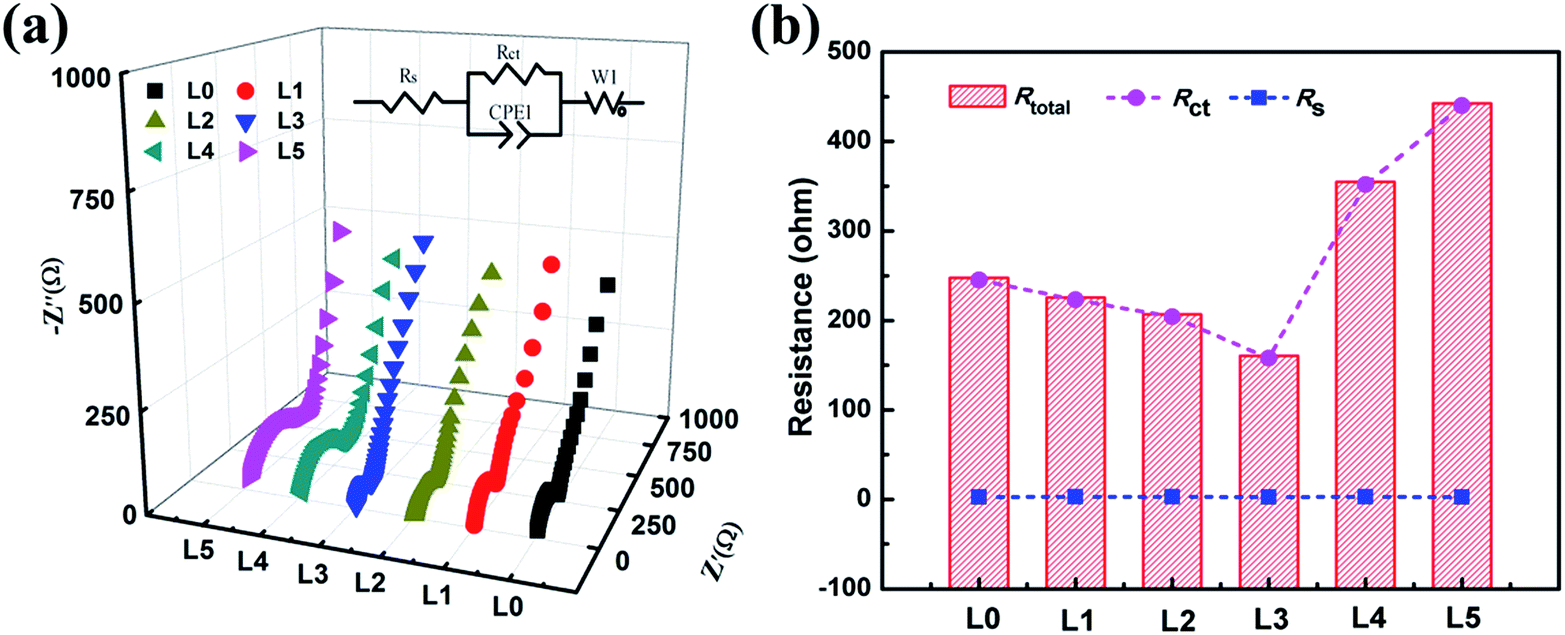 | ||
| Fig. 7 (a) Nyquist plots and the corresponding equivalent circuit (inset) of the samples; (b) histogram of the corresponding fitted parameters of the samples. | ||
4. Conclusions
A novel method is proposed for the first time to synthesize the lithium-rich Li1.5Mn0.75Ni0.25O2.5 material. This method innovatively combines the hydroxide co-precipitation method with the modified molten salt method, in which the co-precipitation method can realize molecule-level mixing of the starting materials and the modified molten salt method, with a moderate excess of 0.5LiNO3–0.5LiOH eutectic salts, can reduce the water waste in the wash process. Different molar ratios (1.55, 1.65, 1.75, 1.85, 1.95, 2.05) of Li/M (M = Ni + Mn) are employed to elucidate the effect of the amount of salt flux on the final product. The structure, morphology and electrochemical performance of these materials were systematically investigated. Structural characterization indicates that the sample with the ratio of Li/M = 1.85 has a more well-defined α-NaFeO2 structure and a more enlarged Li layer spacing. The electrochemical measurements confirm that the Li/M = 1.85 sample has a high initial coulombic efficiency (87.86%), a greatly enhanced rate capability (129 mA h g−1 at 10C) and an excellent cycling stability (98% capacity retention after 50 cycles at 0.2C). Moreover, the EIS results demonstrate that, compared to the other samples, it has a significantly smaller charge transfer resistance (Rct) because of the expansion of the c-parameter and its well-defined α-NaFeO2 layered structure. We anticipate that this novel molten salt method will provide insight into the design and synthesis of lithium-rich materials and should inspire the development of a wide range of other stable, high rate and high capacity cathode materials.Acknowledgements
This work was supported by the Science and Technology Pillar Program of Sichuan University (2014GZ0077), the Development of Advanced Electrode and Electrolytes for LIB (AutoCRC Project 1-111), and the Research Fund for the Doctoral Program of Higher Education, the Ministry of Education (No. 20120181120103).Notes and references
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- A. Manthiram, A. V. Murugan, A. Sarkar and T. Muraliganth, Energy Environ. Sci., 2008, 1, 621 CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- H.-C. Yu, C. Ling, J. Bhattacharya, J. C. Thomas, K. Thornton and A. V. d. Ven, Energy Environ. Sci., 2014, 7, 1760 CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167 CrossRef CAS PubMed.
- C. Yuan, H. B. Wu, Y. Xie and X. W. D. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488 CrossRef CAS PubMed.
- A. Ito, D. Li, Y. Sato, M. Arao, M. Watanabe, M. Hatano, H. Horie and Y. Ohsawa, J. Power Sources, 2010, 195, 567 CrossRef CAS PubMed.
- G. Xu, J. Li, Q. Xue, X. Ren, G. Yan, X. Wang and F. Kang, J. Power Sources, 2014, 248, 894 CrossRef CAS PubMed.
- F. Fu, Y.-P. Deng, C.-H. Shen, G.-L. Xu, X.-X. Peng, Q. Wang, Y.-F. Xu, J.-C. Fang, L. Huang and S.-G. Sun, Electrochem. Commun., 2014, 44, 54 CrossRef CAS PubMed.
- J. Lin, D. Mu, Y. Jin, B. Wu, Y. Ma and F. Wu, J. Power Sources, 2013, 230, 76 CrossRef CAS PubMed.
- F. Cheng, Y. Xin, J. Chen, L. Lu, X. Zhang and H. Zhou, J. Mater. Chem. A, 2013, 1, 5301 CAS.
- L. Zhang, B. Wu, N. Li, D. Mu, C. Zhang and F. Wu, J. Power Sources, 2013, 240, 644 CrossRef CAS PubMed.
- B. Xu, C. R. Fell, M. Chi and Y. S. Meng, Energy Environ. Sci., 2011, 4, 2223 CAS.
- B. Song, H. Liu, Z. Liu, P. Xiao, M. O. Lai and L. Lu, Sci. Rep., 2013, 3, 3094 Search PubMed.
- D. Mohanty, S. Kalnaus, R. A. Meisner, K. J. Rhodes, J. Li, E. A. Payzant, D. L. Wood III and C. Daniel, J. Power Sources, 2013, 229, 239 CrossRef CAS PubMed.
- D. Li, T. Muta and H. Noguchi, J. Power Sources, 2004, 135, 262 CrossRef CAS PubMed.
- T. A. Arunkumar, Y. Wu and A. Manthiram, Chem. Mater., 2007, 19, 3067 CrossRef CAS.
- Y. Jiang, Z. Yang, W. Luo, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, J. Mater. Chem., 2012, 22, 14964 RSC.
- X. He, J. Wang, R. Kloepsch, S. Krueger, H. Jia, H. Liu, B. Vortmann and J. Li, Nano Res., 2014, 7, 110 CrossRef CAS PubMed.
- W. ZhenYao, L. Biao, M. Jin and X. DingGuo, RSC Adv., 2014, 4, 15825 RSC.
- X. Zhao, M. V. Reddy, H. Liu, G. V. S. Rao and B. V. R. Chowdari, RSC Adv., 2014, 4, 24538 RSC.
- Y. Zhao, Y. Sun, Y. Yue, X. Hu and M. Xia, Electrochim. Acta, 2014, 130, 66 CrossRef CAS PubMed.
- C. Yu, G. Li, X. Guan, J. Zheng, L. Li and T. Chen, Electrochim. Acta, 2012, 81, 283 CrossRef CAS PubMed.
- M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, J. Phys. Chem. C, 2007, 111, 11712 CAS.
- M.-H. Lee, Y.-J. Kang, S.-T. Myung and Y.-K. Sun, Electrochim. Acta, 2004, 50, 939 CrossRef CAS PubMed.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112 RSC.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257 RSC.
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS PubMed.
- T. Ohzuku, A. Ueda and M. Nagayama, J. Electrochem. Soc., 1993, 140, 1862 CrossRef CAS PubMed.
- X. Zhang, W. J. Jiang, A. Mauger, Q. Lu, F. Gendron and C. M. Julien, J. Power Sources, 2010, 195, 1292 CrossRef CAS PubMed.
- Z.-D. Huang, X.-M. Liu, S.-W. Oh, B. Zhang, P.-C. Ma and J.-K. Kim, J. Mater. Chem., 2011, 21, 10777 RSC.
- H.-J. Noh, Z. Chen, C. S. Yoon, J. Lu, K. Amine and Y.-K. Sun, Chem. Mater., 2013, 25, 2109 CrossRef CAS.
- K. Amine, H. Tukamoto, H. Yasuda and Y. Fuiita, J. Electrochem. Soc., 1996, 143, 1607 CrossRef CAS PubMed.
- Q. Fu, F. Du, X. Bian, Y. Wang, X. Yan, Y. Zhang, K. Zhu, G. Chen, C. Wang and Y. Wei, J. Mater. Chem. A, 2014, 2, 7555 CAS.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- J. Bareño, M. Balasubramanian, S. H. Kang, J. G. Wen, C. H. Lei, S. V. Pol, I. Petrov and D. P. Abraham, Chem. Mater., 2011, 23, 2039 CrossRef.
- C. Fu, G. Li, D. Luo, J. Zheng and L. Li, J. Mater. Chem. A, 2014, 2, 1471 CAS.
- D. Luo, G. Li, X. Guan, C. Yu, J. Zheng, X. Zhang and L. Li, J. Mater. Chem. A, 2013, 1, 1220 CAS.
- M. G. Kim, M. Jo, Y.-S. Hong and J. Cho, Chem. Commun., 2009, 218 RSC.
- G.-Z. Wei, X. Lu, F.-S. Ke, L. Huang, J.-T. Li, Z.-X. Wang, Z.-Y. Zhou and S.-G. Sun, Adv. Mater., 2010, 22, 4364 CrossRef CAS PubMed.
- Y. Liu, Y. Gao and A. Dou, J. Power Sources, 2014, 248, 679 CrossRef CAS PubMed.
- Q. Li, G. Li, C. Fu, D. Luo, J. Fan and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 10330 CAS.
- S. Wang, Y. Wu, Y. Li, J. Zheng, J. Yang and Y. Yang, Electrochim. Acta, 2014, 133, 100 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06419h |
| This journal is © The Royal Society of Chemistry 2015 |