
Palladium-catalyzed desulfurative Sonogashira cross-coupling reaction of 3-cyano assisted thioamide-type quinolone derivatives with alkynes†
You Wua,
Yongning Xinga,
Jie Wanga,
Qi Sun*a,
Weiqi Konga and
Franck Suzenetb
aState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, PR China. E-mail: sunqi@bjmu.edu.cn; Fax: +86 10 8280 1571; Tel: +86 10 8280 1504
bInstitut of Organic and Analytical Chemistry, University of Orléans UMR-CNRS 7311, rue de chartres, BP 6759, 45067 Orléans Cedex 2, France
First published on 26th May 2015
Abstract
A Pd-catalyzed Cu-mediated desulfurative Sonogashira cross-coupling reaction of thioamide-type quinolone derivatives was proposed for the construction of Csp2–Csp bonds. Alkynylated quinoline derivatives can be easily synthesized in moderate to excellent yields. The mechanism and effect of the 3-cyano on the reaction were also discussed.
A transition metal-catalyzed Sonogashira reaction for the formation of Csp2–Csp bond is an indispensible tool for targeted and parallel synthesis of heterocyclic compounds.1 Traditionally, all of these cross-coupling reactions employ aryl halides, aryl triflates, aryl tosylates and aryl sulfonates as electrophilic coupling partners to perform the cross-coupling with nucleophilic organometallic reagents to produce new C–C bonds. However, the limited stability and/or accessibility of the corresponding heteroaromatic derivatives is somewhat problematic.
In 2000, Liebeskind and Srogl reported a new efficient Pd-catalyzed cross-coupling reaction of thiol ester and thioether species.2 Later, Kappe and co-workers demonstrated that the use of non-basic Liebeskind–Srogl conditions was not only applicable to thiol esters and thioethers as substrates, but could also be extended to latent free thiol group-containing substrates including pyrimidinethiones3 and cyclic thio-amides.4 Considering the wide distribution of C–S bonds in natural products, pesticides and drugs, their transformation via metal-catalyzed coupling reaction have become more and more important in organic chemistry.5
Van der Eycken and co-workers reported an unprecedented microwave-assisted desulfurative Sonogashira cross-coupling protocol for the alkynylation of the C-3 of phenylsulfanylated-2(1H)-pyrazinones.6 It was demonstrated for the first time that the cleavage of C–S from the resin without prior oxidation of the sulfur linker can be realized with the Sonogashira alkynylation of C–S with alkynes in the presence of aryl chloride. The scope of this novel reaction was then extended to pyrimidine functionalized-disulfides.7
Encouraged by Kappe's work, Tatibouët reported the desulfurative alkynylations of 1,3-oxazolidine-2-thiones (OZTs) and 1,3-oxazoline-2-thiones (OXTs) with bases (Scheme 1a, X = O).8 This method was also used in the total synthesis of natural products with anti-tumor activity.9 Hintermann reported a base-free reaction system for the coupling reactions of 2-mercapto-1,3-pyrimidine and alkynes (Scheme 1b), and this method was scoped to 1,3-thiazoline-2-thiones (Scheme 1a, X = S).10 However, the succeeded Sonogashira reactions were only used to substrates of which the latent free thiol functionality linked next to two heteroatoms (marked with red in Scheme 1), such as O, S and N. To the best of our knowledge, Sonogashira coupling reaction of thioamide-type quinolone derivatives with alkynes has not been explored.
 | ||
| Scheme 1 Desulfurative Sonogashira cross-coupling reactions. | ||
Considering the limitation of the scope of the substrates, we inferred that the coordination of the heteroatoms on the substrates, such as oxaline, thiazoline and pyrimidine, to the Pd(0) catalyst could stabilize the Pd(0)-substrate complex to facilitate the oxidative addition procedure, therefore making the cross-coupled products easier to be produced. To simply testify the supposition, we employed 2-mercaptoquinoline (R′ = H, Scheme 1c), which could not provide a heteroatom to coordinate to the Pd(0) catalyst, as the substrate to perform the desulfurative Sonogashira reaction with phenylacetylene. As expected, it failed to afford the desired alkynylated product. In 2011, Sun and co-workers reported a C–H bond activation protocol using cyano as a directing group.11 A mechanism of the stabilizing effect of the coordination between the π-electrons of the cyano group and the Pd(0) species was proposed. Inspired by this work, we designed a 3-cyano-substituted substrate to attempt the desulfurative Sonogashira cross-coupling reaction. To our delight, the desired coupling product was isolated in good yield when 3-cyano-6-methyl-2-mercaptoquinoline 3a was used as the substrate. As a continuing study of the desulfurative Sonogashira cross-coupling reaction, in the present work, we presented a Pd-catalyzed Sonogashira cross-coupling reaction of 3-CN assisted thioamide-type quinolone derivatives with alkynes (Scheme 1c, R′ = CN).
At the beginning of our study, we chose the desulfurative coupling of 3-cyano-6-methyl-2-mercaptoquinoline 3a with phenylacetylene 4a in the presence of the Pd catalyst, Cu additive, CuI and base as a model system to optimize the reaction condition. As shown in Table 1, no reaction occurred without the Pd catalyst (Table 1, entry 1). Among all Pd catalysts including Pd(PPh3)2Cl2, Pd(MeCN)2Cl2, Pd(dba)2, PdCl2, Pd(OAc)2, Pd2(dba)3 and Pd(PPh3)4, Pd(PPh3)4 gave the highest yield under same conditions (Table 1, entries 2–8). No desired product was obtained without the Cu additive, indicating it was essential for the reaction (Table 1, entry 10). CuTC is the optimal Cu additive for this transformation, which gave the expected product in 68% yield (Table 1, entry 9). Reducing the amount of CuI from 1 equiv. to 0.5 equiv. resulted in the same yield. However, a considerable decline of the yield was observed in the absence of CuI, indicating that appropriate amount CuI was necessary for the reaction (Table 1, entries 9, 11 and 12). No desired products were obtained without base and only trace product was obtained without solvent, indicating that they were necessary for the reaction and could significantly affect the reaction (Table 1, entries 18 and 23). Among the bases tested, including Et3N, pyridine, N,N,N′,N′-tetramethylguanidine (TMG), K2CO3, Cs2CO3 and NaOH, Et3N gave the highest yield (Table 1, entries 11 and 13–17). Among all the solvents tested, including THF, DMF, MeCN, CH2Cl2 and dioxane, dioxane gave the highest yield (Table 1, entries 11 and 19–22). The screening of the reaction time indicates that 15 h reaction time is the most suitable to afford the highest yield of product 5aa. A highest yield of 75% was obtained under the optimized conditions as shown (Table 1, entry 26).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Pd (cat.) | “Cu” (1 equiv.) | CuI | Baseb | Solv. | Time (h) | Yieldc (%) |
| a Conditions: 3a (0.5 mmol), 4a (3 equiv.), Pd (Cat.) (0.05 equiv.), “Cu” (1 equiv.), CuI, base (3 mL) and solv. (3 mL) were mixed and heated at 110 °C under argon atmosphere.b Amount of base: TMG (2.5 equiv.), K2CO3 (2.5 equiv.), Cs2CO3 (2.5 equiv.) and NaOH (2.5 equiv.).c Isolated yields. | |||||||
| 1 | — | Cu(OTf)2 | 1 equiv. | Et3N | THF | 20 | 0 |
| 2 | Pd(PPh3)2Cl2 | Cu(OTf)2 | 1 equiv. | Et3N | THF | 20 | 48 |
| 3 | Pd(MeCN)2Cl2 | Cu(OTf)2 | 1 equiv. | Et3N | THF | 20 | 28 |
| 4 | Pd(dba)2 | Cu(OTf)2 | 1 equiv. | Et3N | THF | 20 | 41 |
| 5 | PdCl2 | Cu(OTf)2 | 1 equiv. | Et3N | THF | 20 | 24 |
| 6 | Pd(OAc)2 | Cu(OTf)2 | 1 equiv. | Et3N | THF | 20 | 57 |
| 7 | Pd2(dba)3 | Cu(OTf)2 | 1 equiv. | Et3N | THF | 20 | 30 |
| 8 | Pd(PPh3)4 | Cu(OTf)2 | 1 equiv. | Et3N | THF | 20 | 64 |
| 9 | Pd(PPh3)4 | CuTc | 1 equiv. | Et3N | THF | 20 | 68 |
| 10 | Pd(PPh3)4 | — | 1 equiv. | Et3N | THF | 20 | 0 |
| 11 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | THF | 20 | 68 |
| 12 | Pd(PPh3)4 | CuTc | — | Et3N | THF | 20 | 50 |
| 13 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Pyridine | THF | 20 | 0 |
| 14 | Pd(PPh3)4 | CuTc | 0.5 equiv. | TMG | THF | 20 | 0 |
| 15 | Pd(PPh3)4 | CuTc | 0.5 equiv. | K2CO3 | THF | 20 | 11 |
| 16 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Cs2CO3 | THF | 20 | 60 |
| 17 | Pd(PPh3)4 | CuTc | 0.5 equiv. | NaOH | THF | 20 | Trace |
| 18 | Pd(PPh3)4 | CuTc | 0.5 equiv. | — | THF | 20 | 0 |
| 19 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | DMF | 20 | 14 |
| 20 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | MeCN | 20 | 17 |
| 21 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | CH2Cl2 | 20 | 6 |
| 22 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | Dioxane | 20 | 70 |
| 23 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | — | 20 | Trace |
| 24 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | Dioxane | 5 | 68 |
| 25 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | Dioxane | 10 | 70 |
| 26 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | Dioxane | 15 | 75 |
| 27 | Pd(PPh3)4 | CuTc | 0.5 equiv. | Et3N | Dioxane | 48 | 41 |

The substrate scope of thioamide-type quinolones 3 was then examined under the optimized conditions (Table 2). It is clear that a variety of 3-cyano-2-mercaptoquinolines 3 derivatives can be converted to the desired desulfurative coupling products 5aa–5fa in good yields ranging from 64% to 75%. The reaction can also tolerate well to electron-donating groups such as methyl (5aa, 5ea, 5fa) and methoxy (5da) and electron-withdrawing groups such as fluorine (5ca).

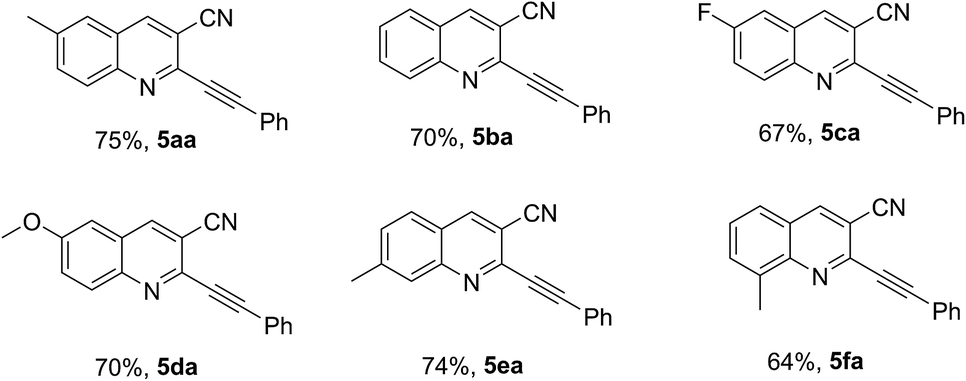
Next, reactions of 3a with various aromatic and aliphatic terminal alkynes were investigated. As shown in Table 3, the reactions with both electron-rich (4b–4e) and electron-poor (4f–4h) aromatic terminal alkynes afforded the corresponding products 5ab–5ah in good yields ranging from 68% to 78%. The reaction with the heteroaryl alkynes afforded the desired products 5ai and 5aj in moderate 46% and 48% yields, respectively. The reactions with aliphatic alkynes 4k and 4m gave the desulfurative coupling products 5ak and 5am in moderate yields of 48% and 56%, respectively, while that with 4l generated the desired product 5al in good yield 78%, possibly due to the steric effect caused by hindering the addition of electrophiles to the C–C triple bond of the product.10,12


The focus of the study was then shifted to screening other substrates applicable to this method that could afford the desired products in higher yields. The reactions of 2-mercapto-3-pyrazine carbonitrile 6a with alkynes occurred highly efficiently (Table 4). The reactions of 6a with electron-rich and electron-poor aryl terminal alkynes produced the desired products in good to excellent yields ranging from 66% to 93% (Table 4, 7aa, 7ab, 7ad and 7af). In addition, the reactions with aliphatic terminal alkynes gave the corresponding products in good to excellent yields ranging from 64% to 91% (Table 4, 7ak, 7al and 7am). The disparity among the yields of 7ak, 7al and 7am once again suggests the significant influence of steric hindrance on the yields of the products. Unexpectedly, the reaction of 3-cyano-2-mercaptopyridine 6b failed to afford the desired product 7ba.


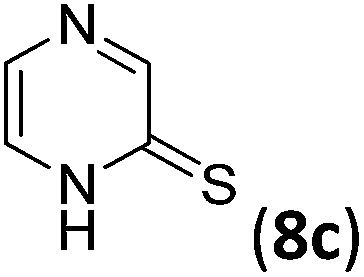
The significant difference between the results of 2-mercaptoquinoline and 3-cyano-2-mercaptoquinolines 3 was evidence that the cyano group crucially facilitated the reaction. Furthermore, a series of experiments were conducted to further verify the effect of the 3-cyano group on the reaction. As mentioned above, the cross-coupling of 3-cyano-2-mercaptoquinoline 3b with phenylacetylene 4a produced the corresponding desulfurative Sonogashira product 5ba in 70% yield (Table 2), while the reaction of 2-mercaptoquinoline 8a with 4a, as shown in Table 5, failed to afford the desired product under the same conditions (Table 5, entry 1). The desired product 9b was isolated in a considerably lower yield of 16% with 3-methyl-2-mercaptoquinoline 8b instead as the substrate (Table 5, entry 2). Likewise, the reaction of 2-mercaptopyrazine 8c afforded the desired product 9c in 20% yield (Table 5, entry 3). However, the reaction of 3-cyano-2-mercaptopyrazine 6a produced the corresponding product 7aa in a significantly higher yield of 88% (Table 4). It was worth mentioning that no desired product was isolated with methyl 3-mercapto-2-pyrazine-2-carboxylate 8d as the substrate. Since the ester group was an electron-withdrawing group, similar to cyano group, it could be inferred that it was the stabilizing coordination effect of the 3-cyano group, other than electron-withdrawing effect, that crucially facilitated the desulfurative Sonogashira cross-coupling reaction.








Based on the mechanism previously reported4 and the discussion above, we proposed a possible mechanism of the critical effect of the 3-cyano group on the reaction (Scheme 2). In the proposed mechanism we highlight the effect of 3-cyano group in transition state C. To a great extent, the reaction undergoes the traditional Liebeskind–Srogl catalytic recycle. Once transition state B8,12 is formed, the activated Pd(0) species coordinates both to the π-electrons of the triple bond of the cyano group and the pyridine ring to produce a stable transition state C,11,13,14 making the oxidative addition of the C–S bond to Pd(0) much easier. This step significantly facilitates the reaction. The transmetalation of C and D leads to the formation of transition state F.8,12 The final product G is afforded by the reductive elimination of Pd, and the Pd(0) species returns to the catalytic recycle.
 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
In summary, we have successfully achieved an efficient 3-cyano assisted desulfurative Sonogashira cross-coupling reaction of thioamide-type quinolone derivatives with Pd(PPh3)4 as the catalyst, readily accessible CuTC and CuI as the additives and cheap Et3N as base. This method can be simply conducted and provide corresponding desulfurative Sonogashira cross-coupling products in moderate to excellent yields. A variety of electron-donating and electron-withdrawing substituent groups are well tolerated. Furthermore, the significant effect of the 3-cyano group, attached next to the thioamide fragment of the substrates, on the Csp2–Csp formation was investigated. Further work on detailed mechanism and extending the application of this method is ongoing in our lab.Acknowledgements
The project is supported by the funds of National Natural Science Foundation of China (NSFC 21272009). We thank Professor Ning Jiao (Peking University) for his review and comments.Notes and references
- Cross-Coupling Reactions in Organic Synthesis, Themed issue, Chem. Soc. Rev., 2011, 40, 1845–2040 Search PubMed.
- L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS.
- A. Lengar and C. O. Kappe, Org. Lett., 2004, 6, 771–774 CrossRef CAS PubMed.
- H. Prokopcová and C. O. Kappe, Adv. Synth. Catal., 2007, 349, 448–452 CrossRef PubMed.
- The reviews about C–S dedulfitative coupling reactions: (a) H. Prokopcová and C. O. Kappe, Angew. Chem., Int. Ed., 2009, 48, 2276–2286 CrossRef PubMed; (b) S. G. Modha, V. P. Mehta and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042–5055 RSC; (c) L. D. Wang, W. He and Z. K. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC; (d) F. Pan and Z.-J. Shi, ACS Catal., 2014, 4, 280–288 CrossRef CAS.
- V. P. Mehta, A. Sharma and E. Van der Eycken, Org. Lett., 2008, 10, 1147–1150 CrossRef CAS PubMed.
- Z.-J. Quan, Y. Lv, F.-Q. Jing, X.-D. Jia, C.-D. Huo and X.-C. Wang, Adv. Synth. Catal., 2014, 356, 325–332 CrossRef CAS PubMed.
- S. Silva, B. Sylla, F. Suzenet, A. Tatibouët, A. Rauter and P. Rollin, Org. Lett., 2008, 10, 853–856 CrossRef CAS PubMed.
- X. Guinchard and E. Roulland, Org. Lett., 2009, 11, 4700–4703 CrossRef CAS PubMed.
- O. V. Maltsev, A. Pöthig and L. Hintermann, Org. Lett., 2014, 16, 1282–1285 CrossRef CAS PubMed.
- W. Li, Z. Xu, P. Sun, X. Jiang and M. Fang, Org. Lett., 2011, 13, 1286–1289 CrossRef CAS PubMed.
- Z.-J. Quan, W.-H. Hu, X.-D. Jia, Z. Zhang, Y.-X. Da and X.-C. Wang, Adv. Synth. Catal., 2012, 354, 2939–2948 CrossRef CAS PubMed.
- W. Li and P. Sun, J. Org. Chem., 2012, 77, 8362–8366 CrossRef CAS PubMed.
- B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786–2791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06337j |
| This journal is © The Royal Society of Chemistry 2015 |