
Highly active Au–Pd nanoparticles supported on three-dimensional graphene–carbon nanotube hybrid for selective oxidation of methanol to methyl formate†
Abstract
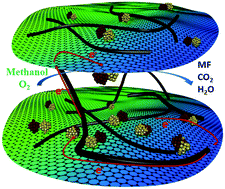
Au–Pd nanoparticles supported on graphene–carbon nanotube hybrid exhibit high catalytic activity in selective oxidation of methanol to methyl formate at low temperature, owning to the strong interaction between graphene and Au–Pd as well as the spacing and bridging effect of nanotube inserter on the hybrid three-dimensional structure.
 Please wait while we load your content...
Please wait while we load your content...

