
Graphene oxide as a corrosion-inhibitive coating on magnesium alloys
Abstract
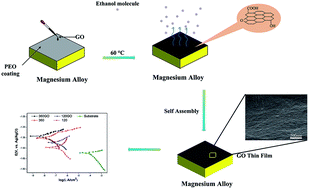
Graphene oxide was formed on the plasma electrolytic oxidation coatings of magnesium alloys via a self-assembly method. The graphene oxide solution was embedded into the porous structures of the coatings to reduce their porosity. The graphene oxide films acted as a physical separation, which inhibited the effects between the protected metal and corrosive media. Furthermore, the film could significantly decrease the corrosion rate compared with pristine magnesium alloy in 3.5 wt% NaCl solution.
 Please wait while we load your content...
Please wait while we load your content...

