
Chemical vapor deposition growth of few-layer graphene for transparent conductive films†
Abstract
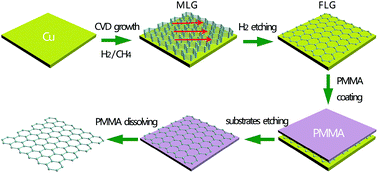
The layer numbers of graphene for graphene based transparent conductive films are crucial. An appropriate number of graphene layers would provide excellent electrical conductivity along with high transparency. Herein, we demonstrated a facile and scalable technique to grow graphene with controllable layers on copper foil substrates using the etching effect of H2 in atmospheric pressure chemical vapor deposition (APCVD), and studied the influence of H2 etching on the properties of graphene transparent conductive films. The etching of formed multi-layer graphene (MLG, 12–14 layers) for Cu substrates assists the formation of few-layer graphene (FLG, 2–3 layers). These as-obtained graphene can be used as high performance transparent conductors, which show improved tradeoff between conductivity and transparency: the transmittance of 96.4% at 550 nm with sheet resistance of ∼360 Ω sq−1, and the transmittance of 86.7% at 550 nm with sheet resistance of ∼142 Ω sq−1. They could be used as high performance transparent conductors in the future.
 Please wait while we load your content...
Please wait while we load your content...

