
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic impact of Pt seed morphology on the highly controlled growth of Ni-based nanostructures†
Carmen
Vázquez-Vázquez
a,
Marcos
Sanlés-Sobrido
a,
Benito
Rodríguez-González
a,
Miguel
Spuch-Calvar
a,
Manuel
Bañobre-López
b,
José
Rivas
b,
Moisés
Pérez-Lorenzo
*a,
Verónica
Salgueiriño
*c and
Miguel A.
Correa-Duarte
*a
aDepartment of Physical Chemistry, Biomedical Research Center (CINBIO), Institute of Biomedical Research of Ourense-Pontevedra-Vigo (IBI), Universidade de Vigo, 36310 Vigo, Spain. E-mail: moisespl@uvigo.es; macorrea@uvigo.es
bInternational Iberian Nanotechnology Laboratory (INL), 4715-330 Braga, Portugal
cDepartment of Applied Physics, Universidade de Vigo, 36310 Vigo, Spain. E-mail: vsalgue@uvigo.es
First published on 8th June 2015
Abstract
A Pt seed-mediated growth method for the synthesis of Ni/NiO and Ni/Ni(OH)2 nanocomposites has been designed so that the morphology and composition of the final materials can be kinetically tailored by a rational choice of the type of seed. Spherical and dendritic Pt nanoparticles provide rather different architectures depending on the different catalytic activity they can exert. Thus, the Pt nanoparticle's initial morphology establishes the conditions for the initial nickel deposition and consequently determines the subsequent autocatalytic growth of the magnetic nanocomposite. Pt seed morphology and reaction conditions reveal the autocatalytic nickel deposition kinetics as a critical factor in the tunability of morphology and composition of nickel-based nanostructures, offering a great opportunity to modulate the final properties of these materials.
Introduction
Colloidal nanoparticles have been widely reported as building blocks for the controlled fabrication of materials with different functionalities. This wide range of capabilities is generally given by the size- and/or shape-dependent properties of the nanoparticles to build such materials.1,2 This fact reflects the importance of colloid chemistry as a synthetic strategy not only to readily control the nanoparticles' properties but also the different features of their assemblies. In this respect, wet-chemistry methods can be used to modulate the processes of nucleation and growth of the nanocrystals and therefore, the nanoparticle size and morphology, which have been demonstrated to be a natural outcome of the way these two stages flow. On this basis, an increasing number of synthetic approaches has been shown to exert an effective control over optical, catalytic, electric and magnetic properties of semiconductor, metallic and metal oxide nanoparticles.1–9 Considering that all these properties stem from the choice of size and shape of these nanostructures, a rigorous understanding of the factors governing nucleation and growth of nanocrystals will pave the way for more rational synthetic approaches, so as to efficiently modulate the properties of the final materials.10The formation of colloidal nanocrystals is generally explained through a mechanism in which the stages of nucleation and growth are widely separated and characterized by different rates. Thus, while the classic LaMer mechanism accounts for a fast, supersaturated-burst nucleation and slow growth,11 the Finke mechanism12 features a slow, continuous nucleation and fast, autocatalytic surface growth. Alternative mechanisms proposed in the literature do not neatly differentiate the time scales for these processes.13,14 In the light of the existing models, the application of a seed-mediated growth method appears as a suitable approach to provide new insights into the general mechanism since it effectively isolates the distinct synthetic steps of nucleation and growth. Given that the fabrication of nanostructures is essentially based on growth phenomena, this segregation enables the assessment of the influence of different factors on the nanocrystal growth kinetics regardless how nuclei are formed. In the end, such evaluation will allow controlling the nanomaterial morphology and therefore, the final properties displayed.
We have positioned this discussion in the frame of the synthesis of nickel-based nanostructures, extensively studied due to their unique electric, magnetic and catalytic properties. Considering that these features are strongly dependent on their morphology,15,16 much interest has been devoted over the last years to synthesize a wide range of architectures based on this metal. As a result, nanorods,16 nanobelts,17 hollow spheres,18 nanocubes,19 nanotubes,20 or nanowires,21 among others, have been fabricated.
We report herein experimental evidence on the key roles that the shape of platinum nanoparticles (PtNPs) used as seeds and different reaction parameters exert on the autocatalytic growth of nickel using nickel–hydrazine complexes as a precursor. To that end, spherical and dendritic PtNPs (from now also referred to as Pt(s) and Pt(d), respectively) were deposited onto different substrates (spherical polystyrene beads (PS, 550 nm diameter), carbon nanotube-wrapped polystyrene spheres (PS/CNT), and anisotropic silica spindles (600 nm length)) enabling the production of nickel-based hybrid nanocomposites with distinct morphologies as well as different Ni/NiO or Ni/Ni(OH)2 ratios. This systematic study will enable us to achieve a better understanding of the mechanisms underlying nickel growth. This is particularly relevant in the implementation of synthetic approaches that afford an accurate control over the structural and compositional parameters of Ni-based nanostructures and therefore, allow the fabrication of materials with precisely tailored properties.
Results and discussion
Ni-based nanostructures can be fabricated using nickel–hydrazine complex precursors in the presence of PtNPs.22–24 The synthetic strategy based on a seed-mediated approach has been modified to tailor the different aspects influencing the growth stage of the process and consequently the final morphology of the Ni-based nanocrystals. With this aim, PtNPs were prepared separately by reduction of potassium tetrachloroplatinate(II) with sodium borohydride and then deposited onto different supports. The synthesis proceeded by the addition of a small volume of different substrate/Pt dispersions to a growth solution containing the nickel precursor, that is, the above-mentioned nickel–hydrazine complexes. These compounds were synthesized by the addition of the appropriate amounts of nickel chloride and hydrazine monohydrate which determine the [N2H4]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Ni2+] molar ratio. Thus, while a [Ni(N2H4)2]Cl2 complex is formed at a molar ratio of [N2H4]:[Ni2+] = 1 or 2, a mixture of [Ni(N2H4)2]Cl2 and [Ni(N2H4)3]Cl2 is produced at a [N2H4]:[Ni2+] = 3 or 4.25,26 Since the [Ni(N2H4)3]Cl2 complex becomes gradually predominant as the [N2H4]:[Ni2+] ratio is increased, the experiments herein described were performed at a [N2H4]:[Ni2+] ≥ 13. It must be pointed out that the nickel reduction process proceeds through these hydrazine precursors that will exhibit different reduction potentials and chemical reactivity. Hence, a pure hydrazine complex was employed not only to avoid additional variables, but also to achieve a more uniform metal reduction and as a result, a more defined crystal growth.
:[Ni2+] molar ratio. Thus, while a [Ni(N2H4)2]Cl2 complex is formed at a molar ratio of [N2H4]:[Ni2+] = 1 or 2, a mixture of [Ni(N2H4)2]Cl2 and [Ni(N2H4)3]Cl2 is produced at a [N2H4]:[Ni2+] = 3 or 4.25,26 Since the [Ni(N2H4)3]Cl2 complex becomes gradually predominant as the [N2H4]:[Ni2+] ratio is increased, the experiments herein described were performed at a [N2H4]:[Ni2+] ≥ 13. It must be pointed out that the nickel reduction process proceeds through these hydrazine precursors that will exhibit different reduction potentials and chemical reactivity. Hence, a pure hydrazine complex was employed not only to avoid additional variables, but also to achieve a more uniform metal reduction and as a result, a more defined crystal growth.
Scheme 1 displays a two-step mechanism for the metallic nickel deposition onto a nanocomposite substrate, beginning with the Pt-catalyzed decomposition of free hydrazine on the platinum seeds27 and followed by an autocatalytic nickel reduction. The first step generates a charged Pt nanoparticle surface, which promotes the reduction of the nickel–hydrazine complex to provide metallic nickel,28 while the second step corresponds to a subsequent growth, controlled and catalyzed by the nickel deposited on every previous step. Since the whole process is carried out in aqueous solution, the final structures always undergo a partial oxidation at the surface. The autocatalytic metallic reduction is indeed an aspect much less considered in the literature, so in order to shed light on this issue, we have employed different approaches to evaluate systematically its impact on the final morphologies obtained. Accordingly, the reduction of the Ni2+ ions included in the nickel–hydrazine complexes has been performed at 40 °C for 2 h in the presence of spherical and dendritic PtNPs (see Fig. S1†) supported on three different substrates: spherical polystyrene beads (PS, 550 nm diameter), carbon nanotube-wrapped polystyrene spheres (PS/CNT) and anisotropic silica spindles (600 nm length).
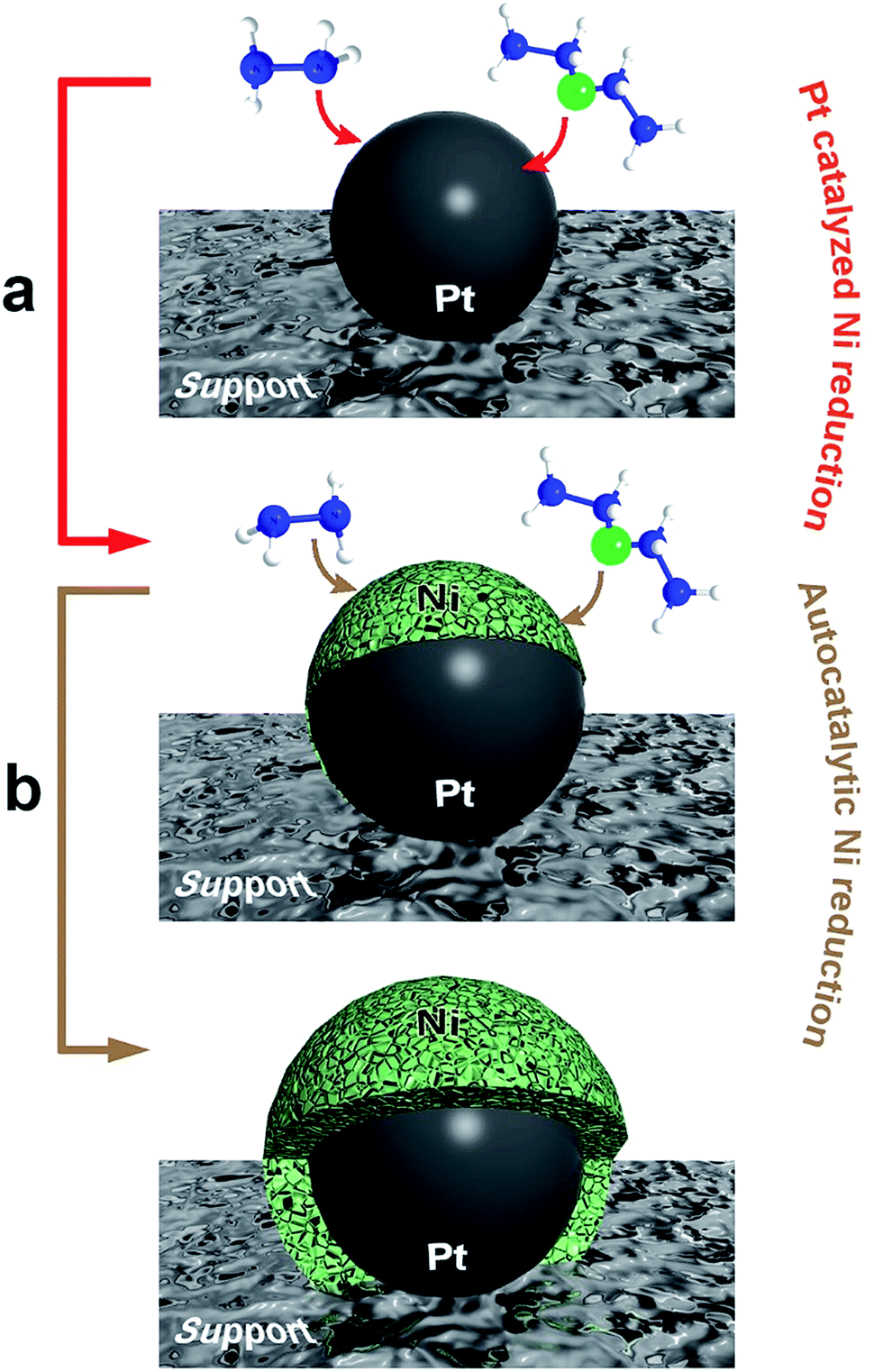 | ||
| Scheme 1 Illustration of the two-stage mechanism for the formation of nickel-based based nanostructures: (a) Pt-catalyzed nickel reduction, and (b) autocatalytic nickel reduction. | ||
Fig. 1 includes the schemes of the substrates employed and the corresponding TEM images of the nickel-based nanostructures obtained onto PS/Pt spheres and CNT/Pt-wrapped PS spheres, through a Pt(s) (left) or Pt(d)-seeded (right) growth. The rather different nature of the substrates employed allows us to consider their influence in terms of both the material (polymer or carbon network) and the radius of curvature (600 or 20 nm average diameter for PS and CNTs, respectively). Fig. 1a and b display the different morphology obtained when using PS beads as supports where either spherical or dendritic Pt seeds have been deposited. As shown, Pt(s) nanoparticles give rise to a sheet-like coating, while Pt(d) seeds lead to a more compact and uniform shell onto the PS spheres. In a similar fashion, the spherical and dendritic PtNPs deposited onto CNTs previously wrapped around PS spheres yield analogous Ni-based architectures (Fig. 1c and d). In view of these results, it seems clear that the nature of the support has no influence on the growth process and the resulting morphology, being the shape of the particles the key factor.
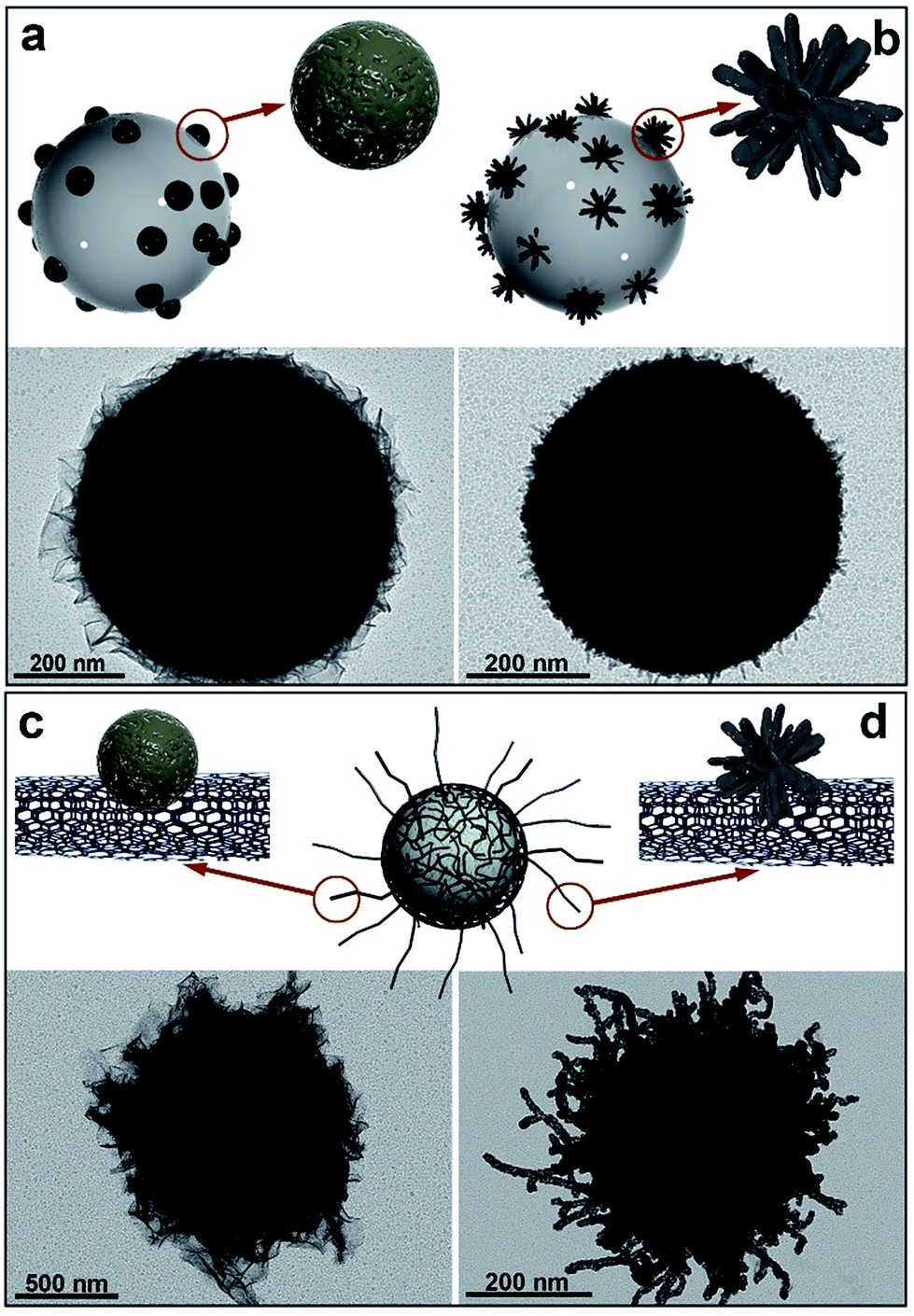 | ||
| Fig. 1 Schemes and TEM images of nickel-based materials grown onto: (a) PS/Pt(s); (b) PS/Pt(d); (c) PS/CNT/Pt(s), and (d) PS/CNT/Pt(d) nanocomposites, at 40 °C for 2 h. [N2H4]:[Ni2+] = 13. | ||
Further evidence on the lack of this effect on the nickel reduction is provided by the use of anisotropic silica spindles where Pt(d) nanoparticles were deposited. These substrates were obtained after coating hematite (α-Fe2O3) nanospindles with silica (Fig. 2a and b). The hematite was then completely removed in a 20% (v/v) hydrochloric acid solution in order to prevent any catalytic interference in the formation on nickel (Fig. 2c). Furthermore, the deposition of the Pt(d) nanoparticles on these anisotropic silica substrates (Fig. 2d) was carried out following the same procedure as for the PS and PS/CNT substrates, such that the nickel reduction and deposition proceed under identical conditions as in the cases previously described. Fig. 2e and f show the TEM and SEM images of the nanostructures obtained, on which uniform and compact Ni-based shells around the silica hollow spindles can be observed.
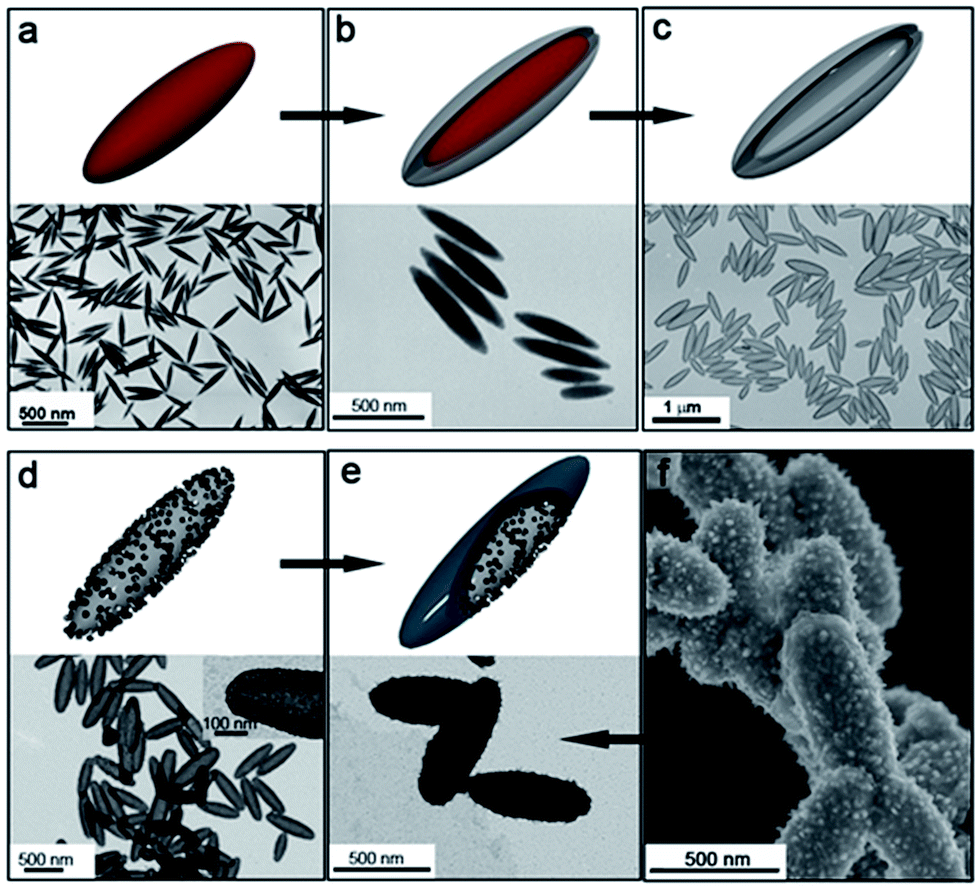 | ||
| Fig. 2 TEM images and schematics of: (a) hematite spindles; (b) silica-coated hematite spindles; (c) hollow silica spindles obtained after removing the iron oxide core; (d) hollow silica spindles after deposition of Pt(d) nanoparticles, and (e) nickel-coated hollow silica spindles after 2 hour growth at 40 °C. (f) SEM image of the nickel-coated hollow silica spindles. | ||
All the experiments described above were performed using the same precursor ([Ni(N2H4)3]Cl2 complex, [N2H4]:[Ni2+] = 13) and reaction conditions (time, temperature and concentrations). Therefore, giving the fact that the substrate exerts no influence, the morphology obtained (either sheets or uniform shells) stems from the different catalytic activity exerted by the Pt seeds. Thus, the process governing the nickel reduction can be considered as driven by the initial Pt-seed mediated growth (Scheme 1a) which plays a shape-directing role along the whole process. Accordingly, the spherical and dendritic PtNPs employed in each case can be considered as templates inducing the initial nickel deposition and therefore, driving the proposed autocatalytic metallic growth.
To shed light on this hypothesis we have focused our attention on the kinetic aspects. The activation energy for the metal reduction onto a preformed seed is considerably lower than that of the reduction of the precursor in a bulk solution.29 Accordingly, it is rational to assume that the formation of the Ni-based nanocomposites herein reported is influenced by the shape-dependent catalytic activity of the pre-existing Pt seeds. Previous results have indeed confirmed this shape-dependent catalytic activity, monitoring the couple ferri/ferrocyanide used as redox probe in the presence of Pt(s) and Pt(d) nanoparticles in a NaBH4 aqueous solution.23 To validate this shape-dependent catalytic activity, both Pt(s) and Pt(d) catalysts were also synthesized using the same reducing and stabilizing agents (NaBH4 and sodium citrate) in order to avoid the introduction of additional variables.
Likewise, both catalysts were supported in 600 nm spherical polystyrene beads, so that other physical phenomena such as a different colloidal stability of the two dispersions can be ruled out and only the morphology of the nanocatalysts plays a role. After conducting the corresponding kinetic analyses, the activation energy (Ea) for the reduction of ferricyanide to ferrocyanide in the presence of PS/Pt(s) and PS/Pt(d) was estimated to be 17.7 ± 1.1 and 12.6 ± 1.2 kJ mol−1, respectively. This lower Ea for the Pt(d)-catalyzed process denotes therefore the key role of structural defect sites commonly found in dendrite-like structures. In fact, atomic under-coordination plays an essential function in promoting electron-transfer reactions since ad-atoms such as steps or kinks are considered to be the real active sites.3 Most importantly, the obtained results point out to the greater efficiency of dendritic PtNPs as reducing catalysts and therefore, to a kinetically favored nickel growth when dendritic seeds are employed. According to these findings, the scenario of the initial reduction rate of the metal precursor becomes largely influenced and consequently, it predetermines the way by which the autocatalytic growth of nickel will evolve. Attending therefore to the morphologies obtained, we can conclude that sheet-like shells obtained result from lower initial reduction rates consequence of Pt(s) seeds, while the uniform compact shells arise at higher rates, typical of Pt(d) nanoparticles, onto both PS or PS/CNTs substrates.
To advance in the understanding of this kinetically controlled metallic growth, the experimental conditions of the synthetic process were modified to increase the reduction rate of the metal precursor and thus, favor metallic nickel deposition. For that reason, the [N2H4]:[Ni2+] ratio as well as the reaction temperature were increased in order to assess the influence of the reduction rate on the final nickel-based architecture. Fig. 3a–c display the evolution over 24 h of the hybrid nanostructures obtained when using a [N2H4]:[Ni2+] = 13 at 40 °C. As shown, the nickel-based nanosheets become larger with the progress of the reaction. Fig. 3d exhibits the hybrid nanocomposites obtained when using a higher [N2H4]:[Ni2+] ratio. In this case, a more uniform and compact nickel-based coating is achieved. A further increase in this molar ratio gives rise to a sharp spike-shaped shell after 2 h (Fig. 3e). Fig. 3f shows the morphology achieved when raising the temperature to 50 °C while keeping [N2H4]:[Ni2+] = 13, characterized by longer and thinner tips. This morphology is in clear contrast with that obtained at the same [N2H4]:[Ni2+] ratio at lower temperature. Interestingly, when similar experiments were performed using the higher catalytically active Pt(d) nanoparticles no sheets or sharp tips were observed (Fig. S2 and S3†) but rather compact shells.
 | ||
| Fig. 3 TEM images of different nickel-based shell morphologies obtained by changing the synthetic conditions using PS/Pt(s) as substrate: (a) [N2H4]:[Ni2+] = 13 at 40 °C for 2 h; (b) [N2H4]:[Ni2+] = 13 at 40 °C for 12 h; (c) [N2H4]:[Ni2+] = 13 at 40 °C for 24 h; (d) [N2H4]:[Ni2+] = 15 at 40 °C for 2 h; (e) [N2H4]:[Ni2+] = 20 at 40 °C for 2 h; (f) [N2H4]:[Ni2+] = 13 at 50 °C for 2 h. | ||
The potential applications of the sheet-like nanostructures observed in Fig. 3a–c, in view of the high specific surface area provided, deserve a comprehensive analysis of the interconnected sheets obtained when using spherical PtNPs. Fig. 4a and b display the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images showing the architectures achieved from the growth of extremely thin curved nanosheets. Fig. 4c shows the STEM energy-dispersive X-ray spectroscopy (STEM-EDS) microanalysis illustrating the location of the Pt, Ni, and O-rich areas of the hybrid nanocomposite. Fig. 4d exhibits the high-resolution transmission electron microscopy (HRTEM) and selected area diffraction (SAED) analyses that reveal the average thickness of the sheets (∼2.3 nm) as well as the face-centered cubic rock salt crystalline structure, characteristic of NiO.
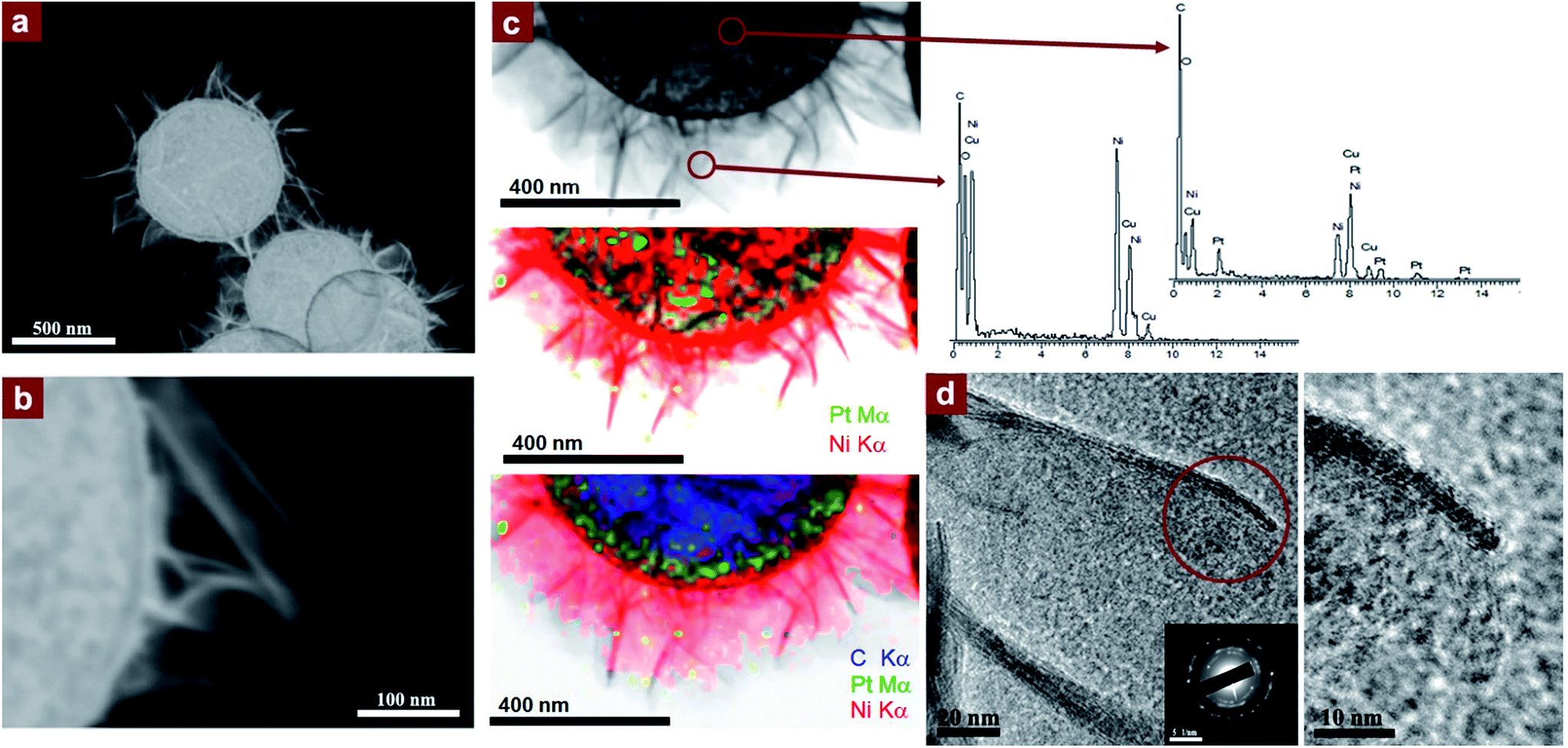 | ||
| Fig. 4 (a and b) HAADF STEM of the nickel-based sheets grown onto the PS/Pt(s) substrates; (c) bright-field STEM images with elemental mappings and EDS microanalyses showing the Pt-rich core and Ni- and O-rich sheet areas; and (d) HRTEM images of the nickel sheets with NiO indexed SAED pattern from the Ni-based nanosheets (inset). | ||
The direct-current (DC) magnetic properties of the different Ni-based nanocomposites were recorded in a SQUID magnetometer and summarized in Fig. 5. These data were collected from five different samples and are displayed in detail in Table 1. Fig. 5a and b display the hysteresis loops collected at 300 K and 5 K, respectively, and Fig. 5c and d include the temperature-dependent magnetization curves, collected in zero-field-cooling (ZFC)-field-cooling (FC) under an applied magnetic field of 100 Oe conditions. The different values of magnetization and coercivity obtained by the samples (see Table 1) can be justified in terms of percentages of metallic Ni and NiO (or Ni(OH)2) and the different diamagnetic supports employed (PS, PS/CNT and hollow silica spindles). Accordingly, the metallic (ferromagnetic) Ni weight fraction decreases as shifting from sample B to E, C, and it is rather low in samples A and D (Table 1 relates the samples to their respective TEM images for easier comparison). One would expect the coercive field values to decrease as the Ni-content increases. This is the case if samples D and E, or A and C are compared, which share the same support (PS) and Pt(s) in the first case, and similar (in terms of magnetic response) supports (PS and silica) and Pt(d) in the second case. However, using the same Pt(d), values of magnetization and coercive field in sample B do not follow these tendencies given its rather different morphology, which implies an increased outer surface due to the carbon nanotubes branching out and consequently, a significant percentage of metallic nickel. Indeed, the outstanding magnetic properties of sample B can be justified on the basis of two main contributions. On one hand, having CNTs as branches spreading out of the spherical substrate increases the available surface where nickel can grow and thus, offers larger quantities of magnetic material in the final composite. On the other hand, the ferromagnetic metallic nickel grown directly onto CNTs has also been reported to improve the magnetic properties of these hybrid materials. This assumption is based on the type of magnetism displayed by C-based units, explained in terms of a mixture of sp2–sp3 carbon atoms that may lead to a ferromagnetic state with an increased magnetization.30
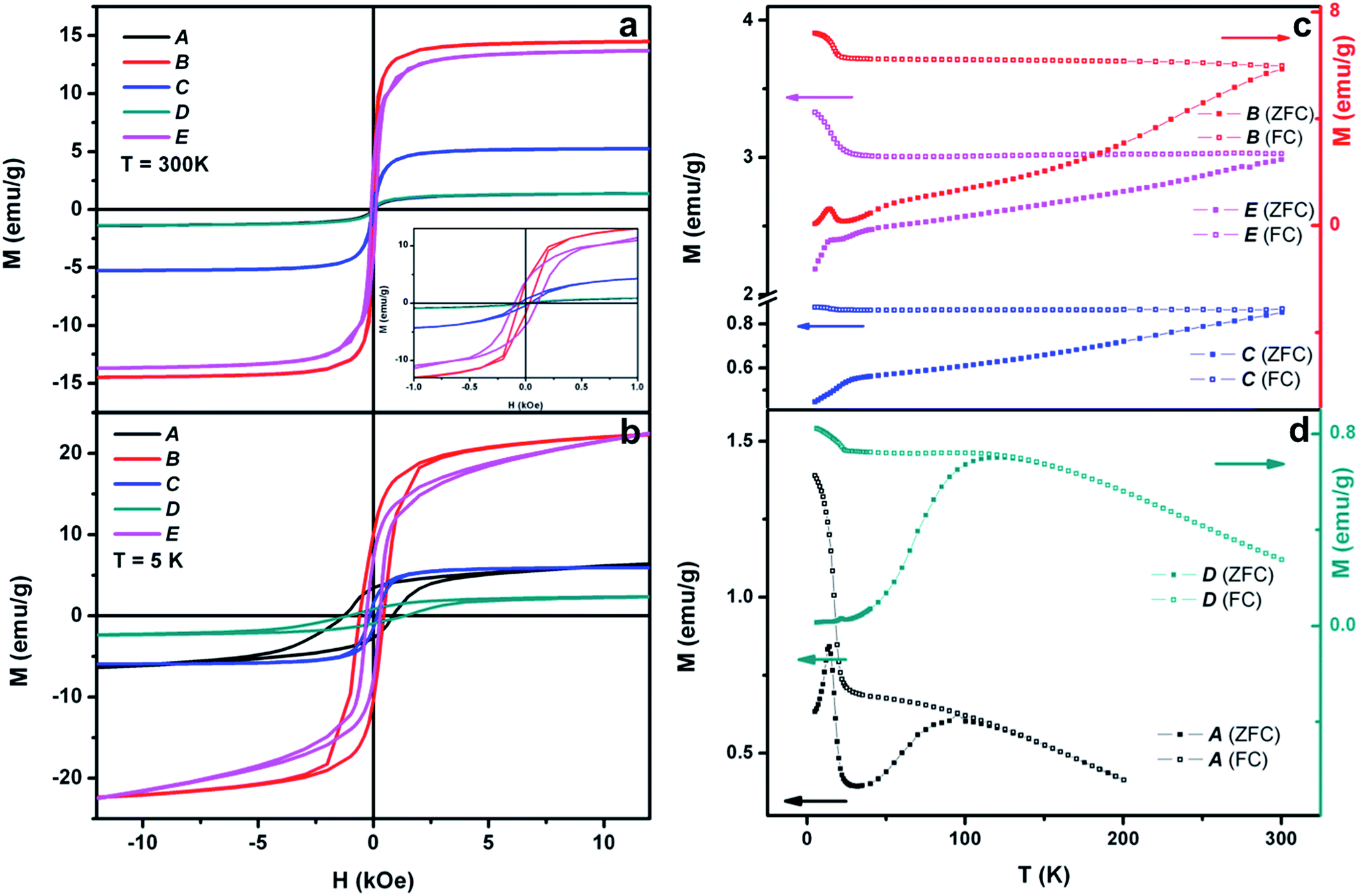 | ||
| Fig. 5 (a and b) Hysteresis loops at 300 K and at 5 K and (c and d) ZFC-FC (100 Oe) temperature-dependent magnetization of the samples labeled as A, B, C, D and E. Note that green and black lines in (a) overlap. | ||
| Sample | Template | TEM image | M s (emu g−1) T = 5 K | M s (emu g−1) T = 300 K | H c (Oe) T = 5 K | H c (Oe) T = 300 K |
|---|---|---|---|---|---|---|
| A | PS/Pt(d) | Fig. 1b | Not saturated | 1.5 | 1286 | — |
| B | PS/CNT/Pt(d) | Fig. 1d | Not saturated | 14.6 | 605 | 90 |
| C | SiO2 spindles/Pt(d) | Fig. 2f | 6.2 | 5.3 | 160 | 54 |
| D | PS/Pt(s) | Fig. 3c | Not saturated | 1.4 | 1118 | — |
| E | PS/Pt(s) | Fig. 3f | Not saturated | 13.8 | 305 | 102 |
The different magnetic phases of the Ni-based nanostructures are better displayed in the ZFC-FC temperature-dependent magnetization curves. Fig. 5b shows this dependence on samples B, C and E, and demonstrates the blocked or ferromagnetic state of the nanostructures due to the important percentage of metallic nickel. Additionally, the peaks at very low temperature reflect the presence of nickel hydroxide (Néel temperature, TN = 23 K) in the three samples. On the other hand, Fig. 5d includes this temperature dependence of samples A and D, which indicates their clear superparamagnetic behavior. Moreover, while sample A M vs. T curves reveal the presence of Ni(OH)2 due to the peak at T = 23 K, sample D evidences the rather small amount of the hydroxide and instead, the presence of the antiferromagnetic NiO (TN = 523 K), as demonstrated by HRTEM.
The overall results reveal therefore a growth process where the combination of Ni-based materials with different composition (and consequently magnetic order) can be kinetically tailored by a rational choice of the seed morphology and the reaction conditions. Thus, while the use of Pt(s) seeds promotes the formation of nanosheets that evolve in size and spread out with the progress of the reaction, the use of Pt(d) seeds promotes the formation of nanoshells which do not change the morphology under the current conditions. Accordingly, the growth observed for the nanosheets may be accounted for by a preferential oxidation of metallic nickel on some facets or more locally on kink or defect site positions of the developing Pt(s)-seeded nickel structure. This anisotropic oxidation consequently restricts the autocatalytic metallic growth in specific crystal directions, which gives rise to a two-dimensional sheet-like architecture of NiO. On the other hand, the faster metallic deposition in the presence of dendritic seeds hinders a preferential anisotropic growth in favor of an isotropic process, providing the compact Pt(d)-seeded nanoshells on which a thin passivated outer layer of Ni(OH)2 prevents any additional metallic nickel growth. The sheet-promoting anisotropic preferential oxidation can be justified in terms of a favored adsorption of oxygen on certain crystalline facets given that this process reduces their surface free energy. Indeed, surface free energies of most metal oxides are invariably lower than the ones corresponding to metal surfaces.31
Subtle changes in the synthetic parameters can also assist in modulating the final architecture of the nanocrystals through the modification of the kinetic behavior. For example, the rise of the reduction rate in the Pt(s) seed-mediated growth by either increasing the [N2H4]:[Ni2+] molar ratio or increasing the reaction temperature, makes available Ni-based nanostructures with different morphologies (Fig. 3). Thus, if the [N2H4]:[Ni2+] ratio is moderately increased, the resulting shape (Fig. 3d) resembles that obtained when using the more catalytically active Pt(d) nanoparticles (Fig. 1b). A further increase in this molar ratio and therefore, in the reduction rate, gives rise to sharp spike-shaped nanoshells (Fig. 3e). A similar growth can also be achieved by raising the temperature while keeping unchanged the [N2H4]:[Ni2+] molar ratio (Fig. 3f). These results contrast to those found in the presence of Pt(d) seeds where changes in the reaction parameters have almost no influence on the final structure of the substrate/Pt/Ni/Ni(OH)2 nanocomposite, which can be explained by the aforementioned passivation in aqueous solution. In any case, the findings herein reported reveal the autocatalytic nickel deposition kinetics and its natural oxidation as the responsible factors for the balance between the isotropic and anisotropic seeded growth of the Ni-based nanostructures as well as the Ni-to-NiO or Ni-to-Ni(OH)2 oxidation.
Conclusions
This study offers new insights into the mechanism of seed-mediated growth of Ni-based nanostructures that allows an accurate control over their morphology and composition and subsequent oxidation. The careful choice of the seed morphology as well as the reaction conditions is of critical importance in the search for synthetic methodologies that permit the fabrication of materials with tailored and well-defined catalytic, magnetic and electronic properties. It is worth highlighting the few nanometers thick NiO sheets and the Ni-based nanostructures with Ni-to-NiO or Ni-to-Ni(OH)2 controlled ratio, which have become particularly important for many applications.Experimental
Unless otherwise noted, all chemicals were purchased from Aldrich and used without further purification.Polystyrene bead functionalization
600 nm polystyrene (PS) beads (Microparticles GmbH, 0.018 standard deviation) were coated with four alternating polyelectrolyte monolayers (PSS/PAH/PSS/PAH) by using an electrostatic self-assembly layer-by-layer protocol. This number was chosen in order to improve the homogeneity of the coating shell and generate the necessary positive surface charge (zeta potential, ξ = +40 mV) for the electrostatic adsorption of 2–3 nm Pt seeds (ξ = −20 mV).CNT functionalization
:1). The sample was then washed with dilute NaOH aqueous solution and then three times with water by centrifugation/redispersion cycles. Finally, the MWNTs were dispersed in water, obtaining a stable dispersion of oxidized MWNTs with carboxylic groups on the walls, providing a negative surface charge.
CNT deposition on the PS surface
Synthesis of anisotropic hollow capsules
Synthesis of platinum seeds and deposition on PS beads, CNTs and anisotropic capsules
:1:0.6 molar ratio. The resulting solution was stirred for 10 min.
Nickel growth
Ni growth onto spherical and dendritic PtNPs supported onto the three different colloidal substrates (PS beads, CNTs, and anisotropic silica capsules) was performed under the same synthetic conditions. As a general approach, a PS/Pt dispersion (0.25 mg mL−1) was added to an aqueous solution (15 mL) containing NiCl2 (0.25 M) and hydrazine (2.5 M). The mixture was kept at 40 °C for 2 h in all cases, then centrifuged (4000 rpm, 15 min) and washed with water and ethanol (twice for each solvent).Characterization
TEM images were obtained using a JEOL JEM 1010 transmission electron microscope operating at an acceleration voltage of 100 kV. HRTEM, STEM and elemental mapping by XEDS analysis were carried out with a JEOL JEM 2010F transmission electron microscope operating at an acceleration voltage of 200 kV. UV-Vis spectra were recorded in a Cary 5000 UV-Vis-NIR spectrophotometer.Acknowledgements
This work was funded by Xunta de Galicia (INBIOMED-FEDER “unha maneira de facer Europa” and EM2014/035), Fundación Ramón Areces, Fundación Tatiana Pérez de Guzmán el Bueno, Spanish Ministerio de Economia y Competividad (CTM2014-58481-R) and European Union Seventh Framework Program [FP7/REGPOT-2012-2013.1] under Grant Agreement no. 316265, BIOCAPS.References
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- M. A. El-Sayed, Acc. Chem. Res., 2004, 37, 326–333 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- J. Hu, Z. Wang and J. Li, Sensors, 2007, 7, 3299–3311 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- M. Sanlés-Sobrido, M. A. Correa-Duarte, S. Carregal-Romero, B. Rodríguez-González, R. A. Álvarez-Puebla, P. Hervés and L. M. Liz-Marzán, Chem. Mater., 2009, 21, 1531–1535 CrossRef.
- R. Narayanan and M. A. El-sayed, J. Phys. Chem. B, 2004, 108, 5726–5733 CrossRef CAS.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS PubMed.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382–10400 CrossRef CAS.
- J.-L. Look, G. H. Bogush and C. F. Zukoski, Faraday Discuss. Chem. Soc., 1990, 90, 345–357 RSC.
- D. G. Duff, P. P. Edwards and B. F. G. Johnson, J. Phys. Chem., 1995, 99, 15934–15944 CrossRef CAS.
- Z. Libor and Q. Zhang, Mater. Chem. Phys., 2009, 114, 902–907 CrossRef CAS PubMed.
- N. Cordente, M. Respaud, F. Senocq, M.-J. Casanove, C. Amiens and B. Chaudret, Nano Lett., 2001, 1, 565–568 CrossRef CAS.
- Z. Liu, S. Li, Y. Yang, S. Peng, Z. Hu and Y. Qian, Adv. Mater., 2003, 15, 1946–1948 CrossRef CAS PubMed.
- J. Bao, Y. Liang, Z. Xu and L. Si, Adv. Mater., 2003, 15, 1832–1835 CrossRef CAS PubMed.
- A. P. LaGrow, B. Ingham, S. Cheong, G. V. M. Williams, C. Dotzler, M. F. Toney, D. A. Jefferson, E. C. Corbos, P. T. Bishop, J. Cookson and R. D. Tilley, J. Am. Chem. Soc., 2011, 134, 855–858 CrossRef PubMed.
- L. Guo, C. Liu, R. Wang, H. Xu, Z. Wu and S. Yang, J. Am. Chem. Soc., 2004, 126, 4530–4531 CrossRef CAS PubMed.
- M. Knez, A. M. Bittner, F. Boes, C. Wege, H. Jeske, E. Maiβ and K. Kern, Nano Lett., 2003, 3, 1079–1082 CrossRef CAS.
- M. Sanlés-Sobrido, M. Bañobre-López, V. Salgueiriño, M. A. Correa-Duarte, B. Rodríguez-González, J. Rivas and L. M. Liz-Marzán, J. Mater. Chem., 2010, 20, 7360–7365 RSC.
- M. Sanlés-Sobrido, M. Pérez-Lorenzo, B. Rodríguez-González, V. Salgueiriño and M. A. Correa-Duarte, Angew. Chem., Int. Ed., 2012, 51, 3877–3882 CrossRef PubMed.
- V. Salgueiriño-Maceira, M. A. Correa-Duarte, M. Bañobre-López, M. Grzelczak, M. Farle, L. M. Liz-Marzán and J. Rivas, Adv. Funct. Mater., 2008, 18, 616–621 CrossRef PubMed.
- G. Huang, S. Xu, G. Xu, L. Li and L. Zhang, Trans. Nonferrous Met. Soc. China, 2009, 19, 389–393 CrossRef CAS.
- J. W. Park, E. H. Chae, S. H. Kim, J. H. Lee, J. W. Kim, S. M. Yoon and J.-Y. Choi, Mater. Chem. Phys., 2006, 97, 371–378 CrossRef CAS PubMed.
- A. V. Anan'ev, M. Y. Boltoeva, N. L. Sukhov, G. L. Bykov and B. G. Ershov, Radiochemistry, 2004, 46, 578–582 CrossRef.
- M. Grzelczak, M. A. Correa-Duarte, V. Salgueiriño-Maceira, B. Rodríguez-González, J. Rivas and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2007, 46, 7026–7030 CrossRef CAS PubMed.
- W. Niu, L. Zhang and G. Xu, Nanoscale, 2013, 5, 3172–3181 RSC.
- K.-H. Han, D. Spemann, P. Esquinazi, R. Höhne, V. Riede and T. Butz, J. Magn. Magn. Mater., 2004, 272–276, 1190–1191 CrossRef CAS PubMed.
- S. H. Overbury, P. A. Bertrand and G. A. Somorjai, Chem. Rev., 1975, 75, 547–560 CrossRef CAS.
- M. A. Correa-Duarte, A. Kosiorek, W. Kandulski, M. Giersig and L. M. Liz-Marzán, Chem. Mater., 2005, 17, 3268–3272 CrossRef CAS.
- M. Sanlés-Sobrido, V. Salgueiriño-Maceira, M. A. Correa-Duarte and L. M. Liz-Marzán, Small, 2008, 4, 583–586 CrossRef PubMed.
- M. Ohmori and E. Matijević, J. Colloid Interface Sci., 1992, 150, 594–598 CrossRef CAS.
- S. Sacanna, L. Rossi, B. W. M. Kuipers and A. P. Philipse, Langmuir, 2006, 22, 1822–1827 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM, HRTEM, BF-STEM and STEM-EDS characterization. See DOI: 10.1039/c5ra05814g |
| This journal is © The Royal Society of Chemistry 2015 |