
Formation of highly porous structure in the electrospun polylactide fibers by swelling-crystallization in poor solvents
Abstract
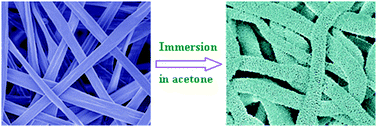
The electrospun fibers made of polylactide (PLA) have been attracting much attention in many applications because of biodegradability and biocompatibility. Introducing porous structure into electrospun PLA fibers is helpful to enhance surface area and thus to extend their applications. Here we report a novel and facile route to produce highly porous structure in the electrospun PLA fibers by simple immersion in poor solvents. It arises from swelling and subsequent solvent-induced crystallization in the electrospun PLA fibers, depending on solvent polarity, immersion temperature and PLA molecular weight. The highly porous PLA fibers have a very large surface area of 137.7 m2 g−1, which is almost unapproachable by other routes.
 Please wait while we load your content...
Please wait while we load your content...

