
Label-free and amplified colorimetric assay of ribonuclease H activity and inhibition based on a novel enzyme-responsive DNAzyme cascade†
Shulan Zenga,
Huakui Huangab,
Yong Huang*ab,
Xiaoqian Liub,
Jian Qinb,
Shulin Zhao*ab,
Zhen-Feng Chena and
Hong Liang*ab
aMinistry of Education Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources, Guangxi Normal University, Guilin, 541004, China. E-mail: huangyong_2009@163.com; zhaoshulin001@163.com; hliang@gxnu.edu.cn
bCollege of Chemistry and Pharmacy, Guangxi Normal University, Guilin, 541004, China
First published on 5th May 2015
Abstract
A simple, label-free and amplified colorimetric assay strategy based on a novel enzyme-responsive DNAzyme cascade has been developed for assay of ribonuclease H activity and inhibition. This novel strategy improved the detection sensitivity by two orders of magnitude over the previously reported methods.
Ribonuclease H (RNase H) is a ribonuclease that can specifically degrade the RNA strand of an RNA–DNA hybrid.1 The specific actions of RNase H are involved in many important biological processes, such as DNA replication, DNA repair, and transcription.2 Moreover, the RNase H activity of HIV-1 reverse transcriptase (RT) is absolutely required for the viral replication cycle, and thus it have been regarded as an attractive target for anti-HIV therapeutics.3 Traditional methods have been established in detecting RNase H activity, such as gel electrophoresis and high-performance liquid chromatography (HPLC).4 However, these methods are laborious, time-consuming, not sensitive, or the necessity of the radiolabeling of the substrate. Many of these limitations are now being addressed by the development of some alternative attractive techniques. For example, fluorescence assays based on the molecular beacon probe, a G-quadruplex formation strategy, and gold nanoparticles (AuNPs) have been developed for detection of RNase H activity.5 Recently, a colorimetric assay using unmodified AuNPs was also used for assay of RNase H activity.6 Although each method has its advantages, the sensitivities of these detection methods are still low due to the lack of powerful signal amplification mechanism. Thus, the development of more sensitive and convenient approaches for RNase H assay is still required.
DNAzymes are in vitro selected catalytic nucleic acids that possess high catalysis activity and specificity toward their substrates similar to protein enzymes.7 More importantly, DNAzymes offer several unique advantages over traditional protein enzymes, such as synthesis convenience, stability against hydrolysis, resistance against denaturation, and relatively lower production costs.8 These important features make DNAzymes excellent biocatalysts for various sensing systems.9 For example, the horseradish peroxidase (HRP)-mimicking DNAzyme was extensively used to amplify detection of metal ions,10 nucleic acids,11 and aptamer substrates.12 Similarly, HRP-mimicking DNAzyme was used for the amplified analysis of T4 polynucleotide kinase, methyltransferase and telomerase.13 Also, metal-dependent DNAzymes were used for the amplified specific sensing of metal ions, nucleic acids, enzyme activity and small organic molecules.14 In addition, the DNAzyme cascade systems were also employed to develop different amplified detection platforms for the quantitative analysis of various types of target molecules.15 However, although numerous DNAzyme-based methods have been reported for the sensing of various targets, no such methods are currently available for RNase H assay.
Herein, we report a simple, label-free and amplified colorimetric assay for RNase H activity and inhibition based on a novel enzyme-responsive DNAzyme cascade. Scheme 1 depicts the design strategy of the DNAzyme cascades for amplified sensing of RNase H. The system consists of a RNA–DNA hybrid hairpin, a G-riched DNA probe, and a blocker DNA. The RNA–DNA hybrid hairpin includes the recognition sites of RNase H in stem section (red) and the Mg2+-dependent DNAzyme (green). The G-riched DNA probe consists of two G-riched DNAzyme segments (blue) and recognition sequence and cleavage site (orange) for the Mg2+-dependent DNAzyme. It is blocked by the blocker DNA (purple) into a quasi-circular structure. Only when RNase H is present, the RNA strand within the RNA–DNA hybrid hairpin is cleaved by RNase H and then generates the activated Mg2+-dependent DNAzyme. The activated Mg2+-dependent DNAzyme can hybridize with a quasi-circular DNA through its complementary arm sequences and then catalyzes the cleavage of the quasi-circular DNA in the presence of cofactor Mg2+, resulting in the release of two G-riched DNAzyme segments and the activated Mg2+-dependent DNAzyme. The released Mg2+-dependent DNAzyme then hybridizes with another quasi-circular DNA to initiate the cleavage of quasi-circular DNA. Eventually, each RNase H-induced activated Mg2+-dependent DNAzyme can go through many cycles, resulting in the cleavage of many quasi-circular DNAs, generating numerous G-riched DNAzyme segments. These generated G-riched DNAzyme segments can recognize and bind with hemin to form the HRP-mimicking DNAzymes, which catalyze the oxidization of ABTS2− (2,2′-azino-bis(3-ethylbenzothiazoline)-6-sulfonate disodium salt) by H2O2 to the colored product ABTS˙− (λmax = 418 nm) that provides the analytical basic of a quantitative measurement of RNase H activity. Remarkably, the use of the dual DNAzyme amplification approach provides significant signal amplification, and substantially improves the sensitivity of colorimetric assay for detecting RNase H. Moreover, this assay does not involve any chemical modification of nucleic acid probes, which is simple and cost-effective.
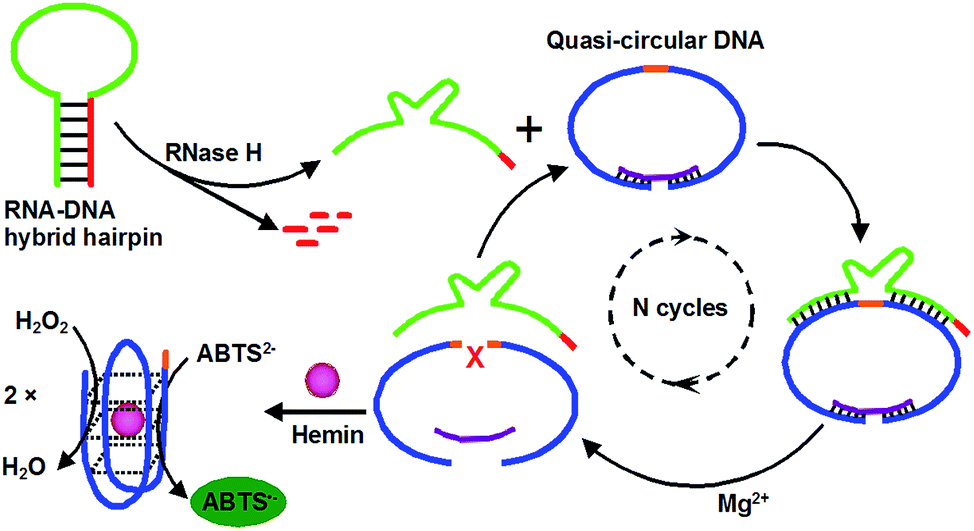 | ||
| Scheme 1 Schematic representation of the RNase H activity assay based on an enzyme-responsive DNAzyme cascade. | ||
To verify the feasibility of the proposed assay strategy, the UV-vis absorption spectra under different conditions were investigated. As shown in Fig. 1, the UV-vis spectrum of hemin only shows low absorbance (418 nm, curve a). This phenomenon was also observed by previous report.12h This absorbance was attributed to the fact that hemin itself had catalytic ability towards the ABTS2−–H2O2 reaction, which had been demonstrated by previous work.12h When mixture of RNA–DNA hybrid hairpin, quasi-circular DNA and hemin in the absence of E. coli RNase H was tested, the absorbance increased slightly (curve b). This might be explained by the fact that the self-hybridization of the hairpin probe could blocked the activation of Mg2+-dependent DNAzyme and HRP-mimicking DNAzyme activity was inhibited by formation of quasi-circular DNA, but still formed the limited HRP-mimicking DNAzymes, which had higher catalytic ability than that of hemin only. Upon addition of E. coli RNase H to the mixture of RNA–DNA hybrid hairpin and quasi-circular DNA, a significant increase of absorbance was observed (curve e), indicating the activation of DNAzyme by RNase H-mediated RNA cleavage, the cleavage of the quasi-circular DNA by the activated DNAzyme and release of G-riched DNAzyme segments. Control experiments revealed that no obvious absorbance change occurred when the predeactivated E. coli RNase H was tested (curve c). These facts demonstrated that the significant absorbance enhancement resulted specifically from E. coli RNase H activity. To further confirm this cleavage and release mechanism, control experiments using Mg2+-dependent DNAzyme and quasi-circularized DNA were also carried out. It was found that the presence of Mg2+-dependent DNAzyme (curve f) caused a significant increase of absorbance compared with that of the blank test (in the absence of Mg2+-dependent DNAzyme, curve d). This results demonstrated that Mg2+-dependent DNAzyme could indeed catalyze the cleavage of quasi-circularized DNA and release of G-riched DNAzyme segments, which bound with hemin to form HRP-mimicking DNAzymes.
 | ||
| Fig. 1 UV-vis absorption spectra of different systems. (a) Hemin only; (b) RNA–DNA hybrid hairpin + quasi-circular DNA + hemin; (c) RNA–DNA hybrid hairpin + quasi-circular DNA + hemin + predeactivated E. coli RNase H; (d) quasi-circular DNA + hemin; (e) RNA–DNA hybrid hairpin + quasi-circular DNA + hemin + E. coli RNase H; (f) quasi-circular DNA + hemin + Mg2+-dependent DNAzyme. | ||
The viability of our designed strategy was further investigated by gel electrophoresis. The experiment results are shown in Fig. 2. The first three lanes represented the RNA–DNA hybrid hairpin probe, the G-riched DNA probe, and the blocker DNA, respectively. Only one bright band appeared in both lane 1 and lane 2, while no bands were observed in lane 3 due to the short single-stranded structure of the blocker DNA. Lane 4 represented the mixture of G-riched DNA probe/blocker DNA mixture, there was a band at almost the same migration position as that in lane 2 due to the short sequences of the blocker DNA. Lane 5 displayed one band for the mixture of the G-riched DNA probe/blocker DNA/RNA–DNA hybrid hairpin, which has the same migration position as that in lane 1 and lane 2, respectively. When RNase H was added to the solution of the RNA–DNA hybrid hairpin probe, the band from the RNA–DNA hybrid hairpin probe disappeared, and a new band with fast mobility was observed (lane 6), indicating the cleavage of the RNA–DNA hybrid hairpin probe by RNase H. Lane 7 represented the mixture of the G-riched DNA probe/blocker DNA/RNA–DNA hybrid hairpin/RNase H. Compared with lane 5, the bands from both RNA–DNA hybrid hairpin probe and mixture of the G-riched DNA probe/blocker DNA disappeared, and two new fast migration bands was observed. This observation demonstrated that the activation of Mg2+-dependent DNAzyme induced by RNase H, and then cleaved G-riched DNA probe/blocker DNA complexes to generate G-riched DNAzyme segments. This was in accordance with the UV-vis scan spectra as shown in Fig. 1. In addition, the circular dichroism (CD) spectra were also used to confirm the cleavage of the quasi-circular DNA, and the generation of G-quadruplex (Fig. S1†).
 | ||
| Fig. 2 Agarose gel electrophoresis analysis: (M) DNA marker; (1) RNA–DNA hybrid hairpin probe only; (2) G-riched DNA probe only; (3) blocker DNA only; (4) mixture of G-riched DNA probe/blocker DNA; (5) the mixture of G-riched DNA probe/blocker DNA complex and RNA–DNA hybrid hairpin probe; (6) the mixture of RNA–DNA hybrid hairpin probe and E. coli RNase H; (7) the mixture of G-riched DNA probe/blocker DNA complex, RNA–DNA hybrid hairpin probe and E. coli RNase H. | ||
To improve the sensitivity of RNase H detection, the ratio of the blocker DNA and the G-riched DNA probe was first optimized. As shown Fig. S2,† the absorbance intensity (at 418 nm) increased gradually with the ratio of the blocker DNA and G-riched DNA probe up to 1.2 and became saturated over 1.2. Thus, the ratio of 1.2 of the blocker DNA and the G-riched DNA probe was selected for the preparation of the quasi-circular DNA. Additionally, the reaction time was also investigated, and the experimental results are shown in Fig. S3.† The absorbance intensity increased rapidly with the reaction time up to 2 h and became slowly after 2 h. By weighing both the sensitivity and the total assay time, the reaction time of 2 h was used for the following experiments.
This DNAzyme-based amplified assay was highly sensitive. Fig. 3 shows the time-dependent absorbance changes upon analyzing different concentrations of E. coli RNase H at the optimal conditions. As the concentration of RNase H increased, the absorbance (at 418 nm) was intensified. This was consistent the enhanced cleavage of hairpin probe by E. coli RNase H, the generation of more activated Mg2+-dependent DNAzymes, the formation of more amounts of G-riched DNAzyme segments and thus the higher amounts of ABTS˙− get. This method enabled the detection of E. coli RNase H as low as 0.001 U mL−1, which was about four orders of magnitude lower than that of the reported AuNP-based colorimetric RNase H assay,6 and two orders of magnitude lower than that of the previous fluorescence methods.5 The high sensitivity of this method was attributed to the reasons as follows: (1) RNase H could catalyze the cleavage of RNA–DNA hybrid hairpin and release Mg2+-dependent DNAzyme units, generating activated Mg2+-dependent DNAzymes. Each of activated Mg2+-dependent DNAzymes could catalyze the cleavage of many quasi-circular DNA substrates via true enzymatic multiple turnovers, releasing many G-riched DNAzyme segments, and thus achieving Mg2+-dependent DNAzyme amplification. (2) The released G-riched DNAzyme segments bound with hemin to form numerous HRP-mimicking DNAzymes. Each HRP-mimicking DNAzyme could also catalyze the oxidization of ABTS2− through true enzymatic multiple turnovers, achieving HRP-mimicking DNAzyme amplification.
 | ||
| Fig. 3 Time-dependent absorbance changes upon analyzing E. coli RNase H by the enzyme-responsive DNAzyme cascade: (a) 0 U mL−1, (b) 0.001 U mL−1, (c) 0.004 U mL−1, (d) 0.02 U mL−1, (e) 0.05 U mL−1, (f) 0.2 U mL−1, (g) 1 U mL−1, (h) 5 U mL−1, and (i) 10 U mL−1. | ||
The proposed DNAzyme-based amplified assay was also specific. To evaluate this property, we challenged the system with the target RNase H and several endonucleases, such as EcoRI, DpnII, EcoRV and BamHI. As shown in Fig. 4, significantly higher absorbance was observed with the target RNase H than with other endonucleases (Fig. 5). These results clearly demonstrate the high specificity of our DNAzyme-based amplified assay for RNase H detection.
 | ||
| Fig. 4 Specificity of the DNAzyme-based amplified assay toward RNase H versus EcoRI, DpnII, EcoRV and BamHI endonucleases. The concentration of RNase H was 0.2 U mL−1, and other endonucleases were 50 U mL−1 each. Error bars were derived from N = 7 experiments. | ||
 | ||
| Fig. 5 Inhibition efficiency of E. coli RNase H activity by ellipticine and oxoglaucine. Error bars were derived from N = 7 experiments. | ||
This simple and sensitive amplified assay was also applied for screening RNase H inhibitors. RNase H inhibitors can prevent the absorbance enhancement by blocking the action of RNase H. The validity of our method in assaying the inhibition of E. coli RNase H activity was first tested by using ellipticine as a model inhibitor. As shown in Fig. 5, as the concentration of ellipticine increased, the inhibition was enhanced (curve a). The IC50 value of ellipticine was found to be 2.65 μM, which are in good agreement with the previous reports.5b Further, the inhibitory effects of oxoglaucine on the E. coli RNase H activity were investigated. It was found that oxoglaucine inhibited E. coli RNase H activity at low concentrations (curve b). The IC50 value for oxoglaucine were found to be 2.5 μM. These results suggested the potentiality of our newly proposed method for screening of RNase H-related potential drugs.
Conclusions
In summary, we have developed a simple, label-free and amplified colorimetric assay for highly sensitive detection of E. coli RNase H based on an enzyme-responsive DNAzyme cascade. This assay technique has several advantages over traditional methods. Firstly, it has very high sensitivity. This assay can detect E. coli RNase H activity as low as 0.001 U mL−1, which is at least two orders of magnitude lower than the previously reported methods. Secondly, in contrast to traditional gel electrophoresis and HPLC methods, the present assay is conducted in aqueous solution, and not requiring separation and troublesome procedures. Thirdly, the nucleic acid probes used here do not require any chemical modification, which is very simple and cost-effective. Finally, this method is based on the absorbance enhancement, therefore, it is amenable to be used in a high-throughput screening format using a multi-well plate reader. Considering these advantages, this approach can be expected to provide a promising tool for sensitive assay of RNase H activity and high-throughput screening of potential drugs.Acknowledgements
This work was supported by the National Natural Science Foundations of China (no. 21305021, 81460544, 21175030), the National Basic Research Program of China (no. 2012CB723501), and the Natural Science Foundations of Guangxi Province (no. 2012GXNSFDA385001, 2013GXNSFBA019038) as well as BAGUI Scholar Program and the project of Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources (Guangxi Normal University), Ministry of Education of China (CMEMR2012-A19, CMEMR2013-C06).Notes and references
- S. J. Schultz and J. J. Champoux, Virus Res., 2008, 134, 86 CrossRef CAS PubMed.
- F. Lazzaro, D. Novarina, F. Amara, D. L. Watt, J. E. Stone, V. Costanzo, P. M. Burgers, T. A. Kunkel, P. Plevani and M. Muzi-Falconi, Mol. Cell, 2012, 45, 99 CrossRef CAS PubMed.
- (a) G. J. Klarmann, M. E. Hawkins and S. F. Le Grice, AIDS Res. Hum. Retroviruses, 2002, 4, 183 Search PubMed; (b) K. Tatyana, E. Francesca, Z. Luca, F. Giovanni, C. Yung-Chi, E. D. Ginger and T. Enzo, Med. Chem., 2009, 5, 398 CrossRef CAS.
- (a) E. Kanaya and S. Kanaya, Eur. J. Biochem., 1995, 231, 557 CrossRef CAS PubMed; (b) S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303 CrossRef CAS PubMed.
- (a) Y. Chen, C. J. Yang, Y. Wu, P. Conlon, Y. Kim, H. Lin and W. Tan, ChemBioChem, 2008, 9, 355 CrossRef CAS PubMed; (b) D. Hu, F. Pu, Z. Huang, J. Ren and X. Qu, Chem.–Eur. J., 2010, 16, 2605 CrossRef CAS PubMed; (c) J. H. Kim, R. A. Estabrook, G. Braun, B. R. Lee and N. O. Reich, Chem. Commun., 2007, 4342 RSC.
- X. Xie, W. Xu, T. Li and X. Liu, Small, 2011, 7, 1393 CrossRef CAS PubMed.
- (a) X. B. Zhang, R. M. Kong and Y. Lu, Annu. Rev. Anal. Chem., 2011, 4, 105 CrossRef CAS PubMed; (b) I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153 RSC.
- (a) T. Fu, X.-H. Zhao, H.-R. Bai, Z.-L. Zhao, R. Hu, R.-M. Kong, X.-B. Zhang, W. Tan and R.-Q. Yu, Chem. Commun., 2013, 49, 664 Search PubMed; (b) J. Liu, A. K. Brown, X. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056 CrossRef CAS PubMed; (c) S. Bi, Y. Yan, S. Hao and S. Zhang, Angew. Chem., Int. Ed., 2010, 49, 4438 CrossRef CAS PubMed; (d) R. Freeman, X. Q. Liu and I. Willner, J. Am. Chem. Soc., 2011, 133, 11597 CrossRef CAS PubMed.
- (a) L. M. Lu, X. B. Zhang, R. M. Kong, B. Yang and W. Tan, J. Am. Chem. Soc., 2011, 133, 11686 CrossRef CAS PubMed; (b) F. Wang, J. Elbaz, C. Teller and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 295 CrossRef CAS PubMed; (c) R. Fu, K. Jeon, C. Jung and H. G. Park, Chem. Commun., 2009, 5838 RSC; (d) T. Li, S. Dong and E. Wang, Chem. Commun., 2007, 4209 RSC; (e) J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2007, 46, 7587 CrossRef CAS PubMed; (f) L. Gong, Z. Zhao, Y.-F. Lv, S.-Y. Huan, T. Fu, X.-B. Zhang, G.-L. Shen and R.-Q. Yu, Chem. Commun., 2015, 51, 979 RSC.
- (a) Y. Hao, Q. Guo, H. Wu, L. Guo, L. Zhong, J. Wang, T. Lin, F. Fu and G. Chen, Biosens. Bioelectron., 2014, 52, 261 CrossRef CAS PubMed; (b) M. A. Aleman-Garcia, R. Orbach and I. Willner, Chem.–Eur. J., 2014, 20, 5619 CrossRef CAS PubMed; (c) C. Ge, Q. Luo, D. Wang, S. Zhao, X. Liang, L. Yu, X. Xing and L. Zeng, Anal. Chem., 2014, 86, 6387 CrossRef CAS PubMed; (d) H. Wang, D. M. Wang, M. X. Gao, J. Wang and C. Z. Huang, Anal. Methods, 2014, 6, 7415 RSC; (e) Y. Yuan, M. Gao, G. Liu, Y. Chai, S. Wei and R. Yuan, Anal. Chim. Acta, 2014, 811, 23 CrossRef CAS PubMed; (f) W. Ren, Y. Zhang, W. T. Huang, N. B. Li and H. Q. Luo, Biosens. Bioelectron., 2015, 68, 266 CrossRef CAS PubMed; (g) G. Pelossof, R. Tel-Vered and I. Willner, Anal. Chem., 2012, 84, 3703 CrossRef CAS PubMed.
- (a) D. Liang, W. You, Y. Yu, Y. Geng, F. Lv and B. Zhang, RSC Adv., 2015, 5, 27571 RSC; (b) K. S. McKeating, D. Graham and K. Faulds, Chem. Commun., 2013, 49, 3206 RSC; (c) L. Shi, Y. Yu, Z. Chen, L. Zhang, S. He, Q. Shi and H. Yang, RSC Adv., 2015, 5, 11541 RSC; (d) P. Zhang, X. Wu, R. Yuan and Y. Chai, Anal. Chem., 2015, 87, 3202 CrossRef CAS PubMed; (e) A. Gomez, N. S. Miller and I. Smolina, Anal. Chem., 2014, 86, 11992 CrossRef CAS PubMed; (f) B. Sai, J. Zhang and S. Zhang, Chem. Commun., 2010, 46, 5509 RSC; (g) S. Bi, M. Chen, X. Jia and Y. Dong, Nanoscale, 2015, 7, 3745 RSC; (h) X. Zhu, H. Zhang, C. Feng, Z. Ye and G. Li, RSC Adv., 2014, 4, 2421 RSC; (i) Y. Gao and B. Li, Anal. Chem., 2014, 86, 888 Search PubMed.
- (a) B. Liu, B. Zhang, G. Chen and D. Tang, Chem. Commun., 2014, 50, 1900 RSC; (b) F. Ma, Y. Yang and C.-Y. Zhang, Anal. Chem., 2014, 86, 6006 CrossRef CAS PubMed; (c) W. Zhou, X. Gong, Y. Xiang, R. Yuan and Y. Chai, Anal. Chem., 2014, 86, 953 CrossRef CAS PubMed; (d) C. Yang, V. Latesc, B. Prieto-Simónd, J.-L. Marty and X. Yang, Talanta, 2013, 116, 520 CrossRef CAS PubMed; (e) H. Mun, E.-J. Jo, T. Li, H.-A. Joung, D.-G. Hong, W.-B. Shim, C. Jung and M.-G. Kim, Biosens. Bioelectron., 2014, 58, 308 CrossRef CAS PubMed; (f) W. Xu, S. Xue, H. Yi, P. Jing, Y. Chai and R. Yuan, Chem. Commun., 2015, 51, 1472 RSC; (g) K. Zhang, T. Ren, K. Wang, X. Zhu, H. Wu and M. Xie, Chem. Commun., 2014, 50, 13342 RSC; (h) Y. Huang, J. Chen, S. Zhao, M. Shi, Z.-F. Chen and H. Liang, Anal. Chem., 2013, 85, 4423 CrossRef CAS PubMed; (i) Y. Zhang and B. Li, Biosens. Bioelectron., 2011, 27, 137 CrossRef CAS PubMed.
- (a) H.-X. Jiang, D.-M. Kong and H.-X. Shen, Biosens. Bioelectron., 2014, 55, 133 CrossRef CAS PubMed; (b) K. He, W. Li, Z. Nie, Y. Huang, Z. Liu, L. Nie and S. Yao, Chem.–Eur. J., 2012, 18, 3992 CrossRef CAS PubMed; (c) C. Jiang, C. Yan, J. Jiang and R. Yu, Anal. Chim. Acta, 2013, 776, 88 CrossRef PubMed; (d) S. Wang, S. Bi, Z. Wang, J. Xia, F. Zhang, M. Yang, R. Gui, Y. Li and Y. Xia, Chem. Commun., 2015, 51, 7927 RSC; (e) H. Wu, K. Zhang, Y. Liu, H. Wang, J. Wu, F. Zhu and P. Zou, Biosens. Bioelectron., 2015, 64, 572 CrossRef CAS PubMed; (f) C. Wang, X. Dong, Q. Liu and K. Wang, Anal. Chim. Acta, 2015, 860, 83 CrossRef CAS PubMed; (g) Y. Zhao, F. Chen, M. Lin and C. Fan, Biosens. Bioelectron., 2014, 54, 565 CrossRef CAS PubMed; (h) Y.-P. Zeng, J. Hu, Y. Long and C.-Y. Zhang, Anal. Chem., 2013, 85, 6143 CrossRef CAS PubMed; (i) H. Li, H.-W. Fu, T. Zhao and D.-M. Kong, RSC Adv., 2015, 5, 6475 RSC; (j) J. Liu, C.-Y. Lu, H. Zhou, J.-J. Xu, Z.-H. Wang and H.-Y. Chen, Chem. Commun., 2013, 49, 6602 RSC.
- (a) P. Wu, K. Hwang, T. Lan and Y. Lu, J. Am. Chem. Soc., 2013, 135, 5254 CrossRef CAS PubMed; (b) W. Zhou, Q. Chen, P.-J. Jimmy Huang, J. Ding and J. Liu, Anal. Chem., 2015, 87, 4001 CrossRef CAS PubMed; (c) L. Zhang, J. Zhao, J. Jiang and R. Yu, Chem. Commun., 2012, 48, 8820 RSC; (d) H. Q. Wang, W. Y. Liu, Z. Wu, L. J. Tang, X. M. Xu, R. Q. Yu and J. H. Jiang, Anal. Chem., 2011, 83, 1883 CrossRef CAS PubMed; (e) H. Wang, M. J. Donovan, L. Meng, Z. Zhao, Y. Kim, M. Ye and W. Tan, Chem.–Eur. J., 2013, 19, 4633 CrossRef CAS PubMed; (f) S. Ye, Y. Guo, J. Xiao and S. Zhang, Chem. Commun., 2013, 49, 3643 RSC; (g) Y. Zhang, L.-J. Wang and C.-Y. Zhang, Chem. Commun., 2014, 50, 1909 RSC; (h) S. Liu, J. Ming, Y. Lin, C. Wang, C. Cheng, T. Liu and L. Wang, Biosens. Bioelectron., 2014, 55, 225 CrossRef CAS PubMed; (i) X.-H. Zhao, L. Gong, X.-B. Zhang, B. Yang, T. Fu, R. Hu, W. Tan and R. Yu, Anal. Chem., 2013, 85, 3614 CrossRef CAS PubMed.
- (a) J. Li, Y. Jia, J. Zheng, W. Zhong, G. Shen, R. Yang and W. Tan, Chem. Commun., 2013, 49, 613 Search PubMed; (b) S. M. Bone, N. J. Hasick, N. E. Lima, S. M. Erskine, E. Mokany and A. V. Todd, Anal. Chem., 2014, 86, 9106 CrossRef CAS PubMed; (c) J. Huang, Y. He, X. Yang, K. Wang, K. Quana and X. Lin, Analyst, 2014, 139, 2994 RSC; (d) T. Tian, H. Xiao, Z. Zhang, Y. Long, S. Peng, S. Wang, X. Zhou, S. Liu and X. Zhou, Chem.–Eur. J., 2013, 19, 92 CrossRef CAS PubMed; (e) L. Gao, L.-L. Li, X. Wang, P. Wu, Y. Cao, B. Liang, X. Li, Y. Lin, Y. Lu and X. Guo, Chem. Sci., 2015, 6, 2469 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, results of CD characterization and results of optimization of assay conditions. See DOI: 10.1039/c5ra05712d |
| This journal is © The Royal Society of Chemistry 2015 |