
Theoretical investigation of H2S removal on γ-Al2O3 surfaces of different hydroxyl coverage†
Rui-Peng Ren,
Xiao-Wei Liu,
Zhi-Jun Zuo* and
Yong-Kang Lv*
Key Laboratory of Coal Science and Technology of Ministry of Education and Shanxi Province, Taiyuan University of Technology, Taiyuan 030024, Shanxi, China. E-mail: zuozhijun@tyut.edu.cn; lykang@tyut.edu.cn; Fax: +86 351 6010386
First published on 3rd June 2015
Abstract
The sulfurized processes of H2S on dehydrated (100) and (110) as well as partially hydrated (110) surfaces of γ-Al2O3 were investigated using a periodic density functional theory method. The adsorption configurations of possible intermediates and the potential energy profiles of reaction are depicted. Our results show that H2S adsorbs preferentially on the Al site along with the S bond, and the adsorption energies are −32.52 and −114.38 kJ mol−1 on the dehydrated (100) and (110) surfaces, respectively. As the reaction temperature of the desulfurization changes, the (110) surface presents different levels of hydroxyl coverage, which affects the adsorption structures of species and reaction energies of dissociation processes. The bonding strengths of H2S on the partially hydrated (110) surfaces are weaker than on the dehydrated (110) surface. Compared with the 3.0 and 8.9 OH per nm2 surfaces, the H2S has the weakest adsorption energy (−39.85 kJ mol−1) and the highest activation energy (92.06 kJ mol−1) on the 5.9 OH per nm2 surface. On the 8.9 OH per nm2 surface, the activation energy of the second dissociation step (rate-determining step) for H2S dissociation is merely 38.32 kJ mol−1. On these involved surfaces, cleavage processes of the two H–S bonds present facile activation energies, which are facilitative to desulfurization.
1. Introduction
Hydrogen sulfide (H2S) is the most common sulfur-containing impurity in combustion of fossil fuels and industrial processes, petroleum/natural gas drilling and refining, coal gasification processes, and biogas generated from anaerobic fermentation of wastes.1–4 It is a prerequisite to reduce the H2S content from these streams to a low level as H2S is extremely malodorous and toxic, being the source of acid rain, causing pipeline corrosion and limiting plant lifetime,5,6 as well as poisoning most downstream catalysts.7Use of solid metal-oxide sorbents, such as CaO, Fe2O3, CuO, ZnO, γ-Al2O3 and CeO2, as candidate desulfurization sorbents has been reported extensively for removal of H2S.8–14 Among these metal oxides, γ-alumina (γ-Al2O3) has attracted considerable attention because of its highly efficient desulfurization capability.15,16 γ-Al2O3 has been widely used in industrial fields, as a catalyst support or as a catalyst (for the Claus process),17 with inevitable reaction with H2S in potential sulfidation environments.18,19 It would be advantageous to understand the detailed reaction process between γ-Al2O3 surface and H2S. Research on this surface reaction may be helpful to improve performance of desulfurizers. Many attempts have been made to investigate the interaction mechanism of H2S with γ-Al2O3, with adsorption of H2S on γ-Al2O3 surface studied using many instruments.15,20–24 For example, Travert et al. studied interaction of H2S on γ-Al2O3 surface using infrared (IR) spectroscopy,25 and showed that the adsorption of H2S irreversibly perturbs the high-frequency region of the IR spectroscopy and results in changes to surface acidity. DeRosset et al.26 estimated that isosteric heats of adsorption of H2S ranged from −104.6 to −159.0 kJ mol−1, depending on the degree of dehydration of the γ-Al2O3. And entropy calculations indicate that mobility of the adsorbed H2S is highly restricted. Reshetenko et al.16 carried out heterogeneous decomposition of H2S on γ-Al2O3, and determined the reaction order and effective activation energy to be 2.0 and 72 kJ mol−1, respectively.
Recently, the density functional theory (DFT) method based on quantum chemistry has been proposed as a method for providing some molecular and atomic level information regarding the γ-Al2O3 surface, including the positions and behaviors of sulfur components. Arrouvel et al.27 investigated the interaction between H2S and the γ-Al2O3 surface under the usual hydrodesulfurization (HDS) conditions using DFT combined with surface thermochemistry. They pointed out that only rather high temperatures and very low water partial pressure stabilize the sulfidation of the (110) surface, leading to formation of sulfhydryls and hydroxyls. Lo et al.28 also studied adsorption of H2S on γ-Al2O3 surfaces, including both dehydrated and hydrated surfaces. They found that the chemisorption of H2S on dehydrated surfaces is highly favored, whereas the phenomenon of physisorption is more likely to occur on the hydroxyl surfaces. Furthermore, H2S adsorption on dehydrated surfaces is more energetically favorable than on hydrated surfaces. Although the adsorption properties of H2S on γ-Al2O3 surfaces have been characterized experimentally and theoretically, a detailed sulfurized mechanism has not been reported to date.
In this study, based on DFT together with a periodic model, we systematically investigate the adsorption energies and geometries of H2S and resultant species on different γ-Al2O3 surfaces, including partially hydrated (110), and dehydrated (100) and (110) surfaces. The reaction processes and potential energy surfaces of H2S decomposition on the different surfaces are also calculated, which may aid in understanding the reaction state of H2S under different operating environments and in developing new-style desulfurizers.
2. Computational methods and models
2.1. Calculation methods
All Kohn–Sham DFT calculations were performed using a periodic model and a plane-wave basis set, as implemented in the Vienna ab initio simulation package (VASP).29,30 The interaction between valence electrons and the core was described by the full-potential projector augmented wave (PAW) method.31,32 The generalized gradient approximation (GGA) formulation of Perdew, Burke and Enzerhoff (PBE)33 was employed to treat exchange–correlation energy. Brillouin zone integration was converged with a 3 × 3 × 1 k-point mesh generated by the Monkhorst–Pack algorithm.34 Previous calculations have shown that the 3 × 3 × 1 k-point mesh is sufficient to gain good converged results.35,36 We studied its reliability (see Table S1 in ESI†) and found that the adsorption energies and activation energy are similar. Therefore, we used the 3 × 3 × 1 k-point mesh. A cutoff energy of 400 eV was sufficient to obtain a satisfactory convergence of the total energy. In the structure optimization and energy calculation, the convergence tolerance was set to 10−5 eV for electronic self-consistent iteration and the residual forces of free atoms were limited smaller than 0.03 eV Å−1. A Gaussian smearing function with a width of 0.1 eV was utilized to speed up convergence of the total energy. Spin polarization was used in all calculations. The transition states (TSs) and the minimum energy paths (MEPs) were located using the nudged elastic band (NEB) method.37,38 Eight equally spaced images were linearly interpolated between the reactant and final states. The quasi-Newton algorithm was used to relax the ion positions in all NEB calculations. All reported transition structures were verified to exhibit only one imaginary frequency by frequency calculations.2.2. Surface models
The crystallographic bulk structure of γ-Al2O3 is complex and controversial because of the diversity of metastable phases during the preparation process. As far as we know, three typical γ-Al2O3 structures have been reported: the traditional defective spinel structure,39 the Paglia structure,40 and the Digne structure.41 In particular, the Digne structure is widely used in industrial catalysis applications.42–44 Thus, the Digne structure of γ-Al2O3 was chosen for this study. The (110) and (100) surfaces are the main surfaces of γ-Al2O3, and contribute to approximately 90% of the overall surface area.45 In the medium-high temperature desulfurization environment,46,47 the two surfaces are shown to have different coverage of surface hydroxyls. According to a report by Digne et al.,45 the (100) surface is fully dehydrated above the Claus reaction conditions(∼600 K), and the (110) surface contains 8.9 OH per nm2, 5.9 OH per nm2, or 3.0 OH per nm2 in the temperature range from 600 K to 1150 K. Therefore, the present study focuses on dehydrated (100) and (110) surfaces, and partially hydrated (110) surfaces including 3.0 OH per nm2, 5.9 OH per nm2, and 8.9 OH per nm2 surfaces. The (100) and (110) surfaces were modeled using a p(2 × 1) supercell and a p(1 × 1) supercell, respectively. For the (100) surface, a four-layer slab was employed, where the two surface layers were relaxed and the bottom two layers were kept fixed in their bulk position. For the (110) surface, the slab consisted of five atomic layers, with the bottom two layers frozen to the bulk parameters and the remaining layers allowed to relax. In all calculations, adsorbates were placed on one side of the slab, and a 15 Å thick vacuum spacer was placed in the perpendicular direction to separate the surface slab.The adsorption energy, Ead, was defined as
| Ead = E(ads/slab) − E(slab) − E(ads) |
The reaction energy (ΔE) and the activation energy (Ea) are defined as
| ΔE = E(FS) − E(R) |
| Ea = E(TS) − E(R) |
3. Results and discussion
3.1. Bulk γ-Al2O3 and gas-phase H2S and HS in vacuum
In the present study, the bulk structure of γ-Al2O3 is monoclinic, and the unit cell contains 8 Al2O3 units. The cell volume after structure optimization is 369.13 Å3 (46.14 Å3 per Al2O3 unit, namely, the cell volume per Al2O3 unit), which is about 0.54% smaller than the experimental value (46.39 Å3 per Al2O3 unit).48 The corresponding lattice parameters after geometry optimization are a = 5.52 Å, b = 8.33 Å, c = 8.02 Å, and β = 90.62°. The largest deviation of these values is slightly less (1.08%) than the experimental value reported by Krokidis et al.49 The relevant results of gas-phase H2S and HS in vacuum, including bond lengths, bond angles, and vibrational frequencies, are summarized in Table 1. These calculated values are consistent with previous experimental and calculational data.50–523.2. Adsorption geometries and energies on the γ-Al2O3 surfaces
Fig. 1 shows the stable configurations of the dehydrated model γ-Al2O3 (100) and (110) surfaces, and the potential adsorption sites, which are used to investigate the interaction with possible adsorbates. For the purposes of discussion, the Al and O atoms in the outmost layer of the slab are labeled I, II, III, IV and A, B, C, D, respectively. On the dehydrated γ-Al2O3 (100) surface, Al(I)–Al(III) exposed on the surface are pentacoordinated, whereas Al(IV) is tetracoordinated and in a position below the surface plane which is not available for adsorption.53 All the O atoms are tricoordinated and labeled for the different marks because of different chemical environments. In the case of the dehydrated γ-Al2O3 (110) surface, the Al(I) and Al(II) atoms are tetracoordinated but have different chemical environments, and Al(III) is tricoordinated. The O(A) and O(B) atoms are tricoordinated, and the O(C) and O(D) atoms are dicoordinated. The geometries of the partially hydrated (110) surfaces are shown in Fig. 2, including the 3.0, 5.9, and 8.9 OH per nm2 surfaces. In particular, the sites on the partially hydrated (110) surfaces share the marks of the dehydrated surface. | ||
| Fig. 1 Stable configurations of the dehydrated model γ-Al2O3 (100) and (110) surfaces: (a) (100) surface; (b) (110) surface. The I, II, III, and IV labels refer to the Al sites, and A, B, C, and D stand for the O sites (gray, Al; red, O). | ||
 | ||
| Fig. 2 Stable configurations of the model γ-Al2O3 (110) surfaces for different hydroxyl coverage: hydrated with θ = 3.0 OH per nm2; hydrated with θ = 5.9 OH per nm2; hydrated with θ = 8.9 OH per nm2 (gray, Al; red, O; white, H). | ||
 | ||
| Fig. 3 Optimized geometric structures of H2S, HS, S, and H adsorbed on the dehydrated γ-Al2O3 (100) surface. Distances are given in Å (gray, Al; red, O; yellow, S; white, H). | ||
| Species | Parama | D100b | D110c | 3.0 OHe per nm2 | 5.9 OH per nm2 | 8.9 OH per nm2 |
|---|---|---|---|---|---|---|
| a Param: parameters.b D100, the dehydrated γ-Al2O3 (100) surface.c D110, the dehydrated γ-Al2O3 (110) surface.d FS denotes the final state.e 3.0 OH per nm2, 5.9 OH per nm2 and 8.9 OH per nm2 represent the different levels of hydroxyl coverage for γ-Al2O3 (110) surface. | ||||||
| H2S | Site | Al III | Al III | Al II | Al I | Al I |
| dH–S (Å) | 1.354/1.353 | 1.454/1.355 | 1.380/1.370 | 1.396/1.352 | 1.416/1.362 | |
| dS–Al (Å) | 2.657 | 2.417 | 2.430 | 2.586 | 2.540 | |
| ∠HSH (deg) | 91.0 | 95.0 | 90.6 | 94.7 | 94.1 | |
| Ead (kJ mol−1) | −32.52 | −114.38 | −92.82 | −39.85 | −67.57 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| HS | Site | Al III | Al III | Al I–I | Al I–I | Al I |
| dH–S (Å) | 1.354 | 1.354 | 1.352 | 1.352 | 1.366 | |
| dS–Al (Å) | 2.425 | 2.284 | 2.425/2.477 | 2.405/2.540 | 2.383 | |
| Ead (kJ mol−1) | −96.48 | −208.75 | −154.79 | −158.64 | −155.06 | |
|
||||||
| S | Site | Al III-OD | Al III-OC/Al I-OB | Al I-OB | Al I-OB | OA |
| dS–O (Å) | 1.788 | 1.765/1.741 | 1.739 | 1.738 | 1.812 | |
| dS–Al (Å) | 2.388 | 2.263/2.224 | 2.235 | 2.223 | — | |
| Ead (kJ mol−1) | −256.26 | −308.75/−318.59 | −318.41 | −307.44 | −258.12 | |
|
||||||
| H | Site | OC/OD | OC | OC | OA | OA |
| dS–H (Å) | 0.983/0.981 | 1.091 | 0.979 | 0.972 | 0.971 | |
| Ead (kJ mol−1) | −143.49/−143.55 | −340.80 | −269.73 | −268.15 | −273.33 | |
| HS + H | Ead (kJ mol−1) | −587.55(FS1a)d | −672.14(FS1c) | −640.40(FS1e) | −619.84(FS1g) | −636.32(FS1i) |
| S + H | Ead (kJ mol−1) | −652.14(FS2b) | −798.68(FS2d) | −734.67(FS2f) | −712.82(FS2h) | −687.67(FS2j) |
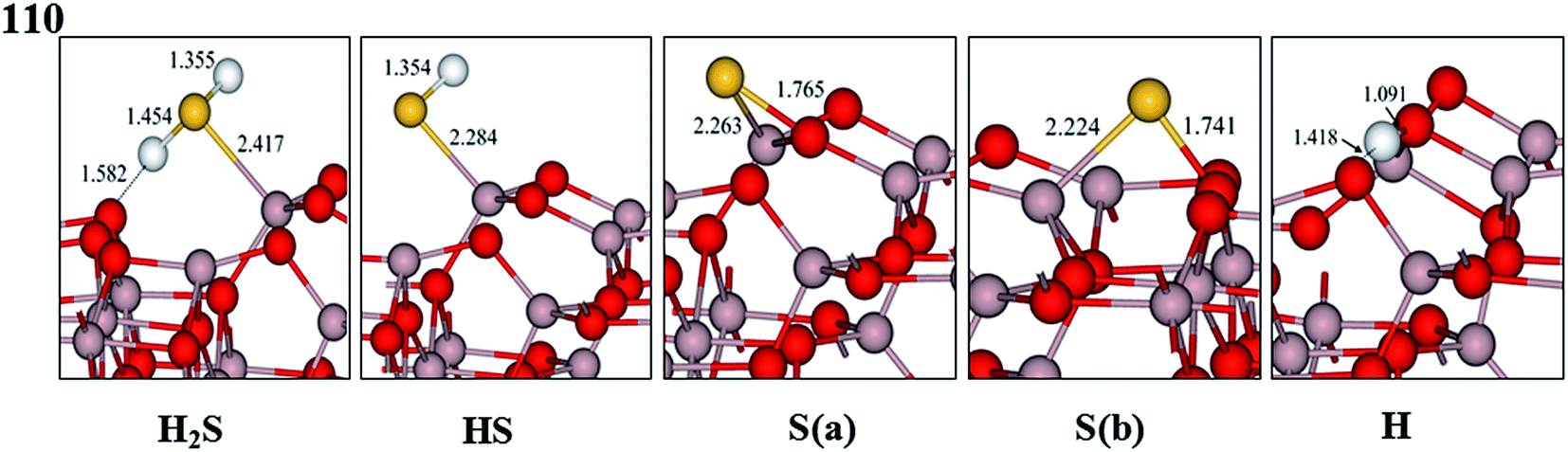 | ||
| Fig. 4 Optimized geometric structures of H2S, HS, S, and H adsorbed on the dehydrated γ-Al2O3 (110) surface. Distances are given in Å (gray, Al; red, O; yellow, S; white, H). | ||
 | ||
| Fig. 5 Optimized geometric structures of H2S, HS, S, and H adsorbed on the partially hydrated γ-Al2O3 (110) surfaces. Distances are given in Å (gray, Al; red, O; yellow, S; white, H). | ||
In terms of HS adsorption on the 3.0 OH per nm2 surface, the HS steadily absorbs on two adjacent Al(I) atoms via the bridge bond mode (see Fig. 5 HS), and the corresponding adsorption energy is −154.79 kJ mol−1. Similar to HS adsorption on the 3.0 OH per nm2 surface, the S atom of HS is in direct contact with both Al(I) sites on the 5.9 OH per nm2 surface. However, the bridge site occupied by the HS favorably absorbs on the single Al(I) site and its H atom lies toward the O(A) site on the 8.9 OH per nm2 surface.
For S adsorption on the 3.0 OH per nm2 surface, the S atom binds to the surface Al(I) and O(B) atoms via the bridge bond mode forming a stable structure, as shown in Fig. 5, with absorption energy of −318.41 kJ mol−1. Likewise, the S atom that binds with the Al(I)–O(B) bridge site is still stable on the 5.9 OH per nm2 surface with adsorption energies of −307.44 kJ mol−1, and the bond lengths of S–Al(I) and S–O(B) are 2.223 and 1.738 Å, respectively. In contrast with the 3.0 OH per nm2 and 5.9 OH per nm2 surfaces, the S atom preferentially occupies the O(A) site with a S bond on the 8.9 OH per nm2 surface with an absorption energy of −258.12 kJ mol−1, which is 57.03 kJ mol−1 lower than that on the Al(I)–O(B) bridge site. The adsorbed surface hydroxyl inhibits S adsorption on the Al(I)–O(B) bridge site.
For H adsorption, the optimal adsorption sites are O(C) on the 3.0 OH per nm2 surface, and O(A) on the 5.9 and 8.9 OH per nm2 surfaces, as illustrated in Fig. 5, and the corresponding adsorption energies are −269.73, −268.15, and −273.33 kJ mol−1, respectively. These absorption energy values are approximately 70 kJ mol−1 higher compared with the dehydrated surface. This indicates that the bonding strength of the H adsorption is weaker than on the dehydrated surface.
3.3. Reaction mechanism of H2S/γ-Al2O3 interactions
The probable potential energy profiles of H2S interacting with the γ-Al2O3 surfaces are shown in Fig. 6, and the calculated values of reaction energy and activation energy for each elementary step are given. Fig. 6(a) shows the first dehydrogenation process (H2S → HS + H), and the second dehydrogenation step (HS → H + S) is displayed in Fig. 6(b). The relative structures on this potential energy profiles are depicted in Fig. 7–9, which include final states (FSs) and TSs on the different γ-Al2O3 surfaces. In addition, the coadsorption energies of FSs are given in the Table 2. In particular, the reaction of H2S on hydrated (110) surfaces will never involve desorption of H2O prior to S–H activation because of the high adsorption energy of H2O, similar to the CH4 dissociation on the hydrated γ-Al2O3 surfaces.44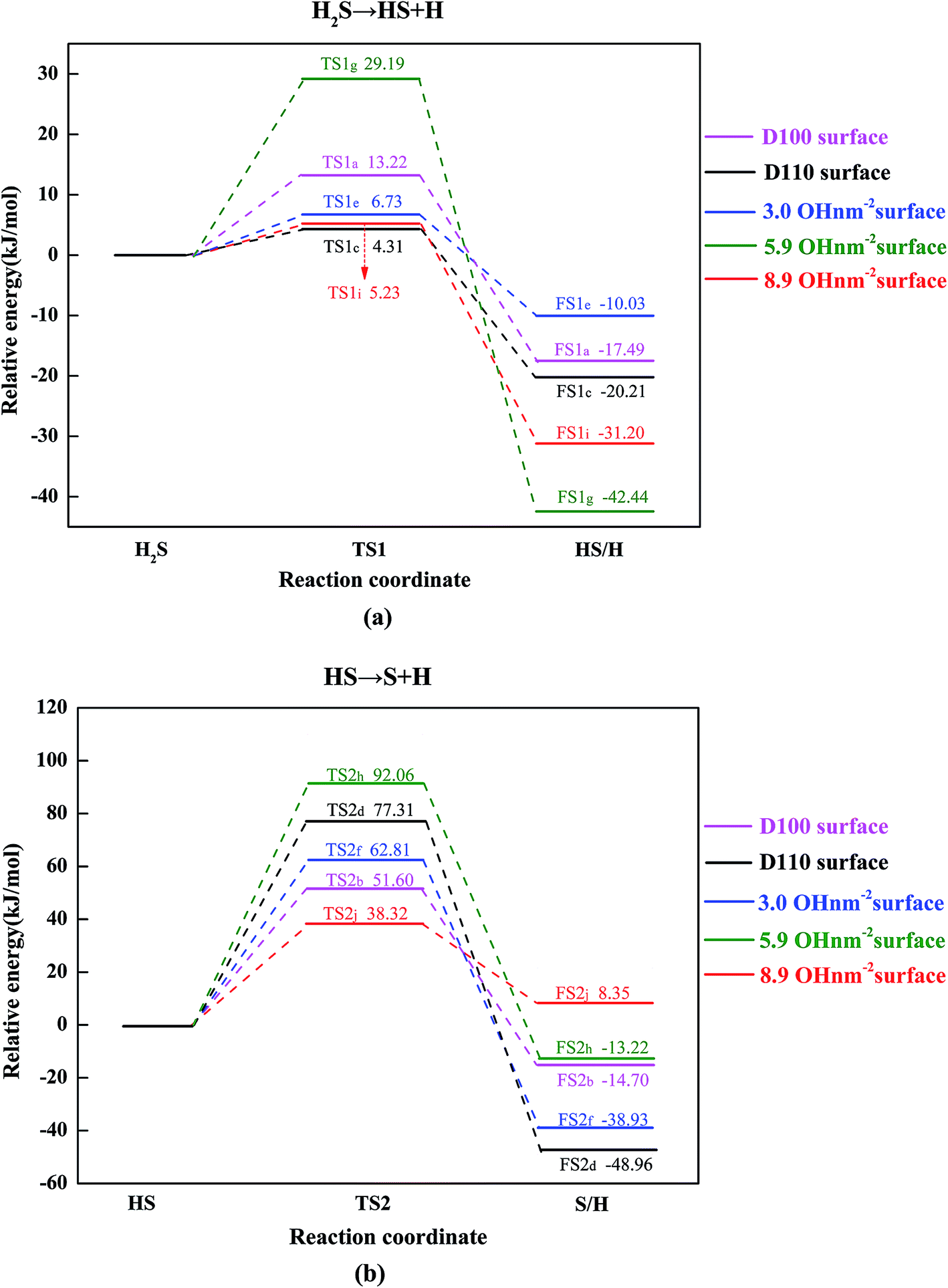 | ||
| Fig. 6 Calculated probable potential energy profiles for dissociation of H2S (a) and HS (b) on dehydrated and partially hydrated γ-Al2O3 surfaces. (D100 surface, the dehydrated γ-Al2O3 (100) surface; D110 surface, the dehydrated γ-Al2O3 (110) surface). | ||
 | ||
| Fig. 7 Calculated transition states (TSs) and corresponding final states (FSs) for dissociation of H2S and HS on the dehydrated γ-Al2O3 (100) surface. Distances are given in Å (gray, Al; red, O; yellow, S; white, H). | ||
 | ||
| Fig. 8 Calculated transition states (TSs) and corresponding final states (FSs) for dissociation of H2S and HS on the dehydrated γ-Al2O3 (110) surface. Distances are given in Å (gray, Al; red, O; yellow, S; white, H). | ||
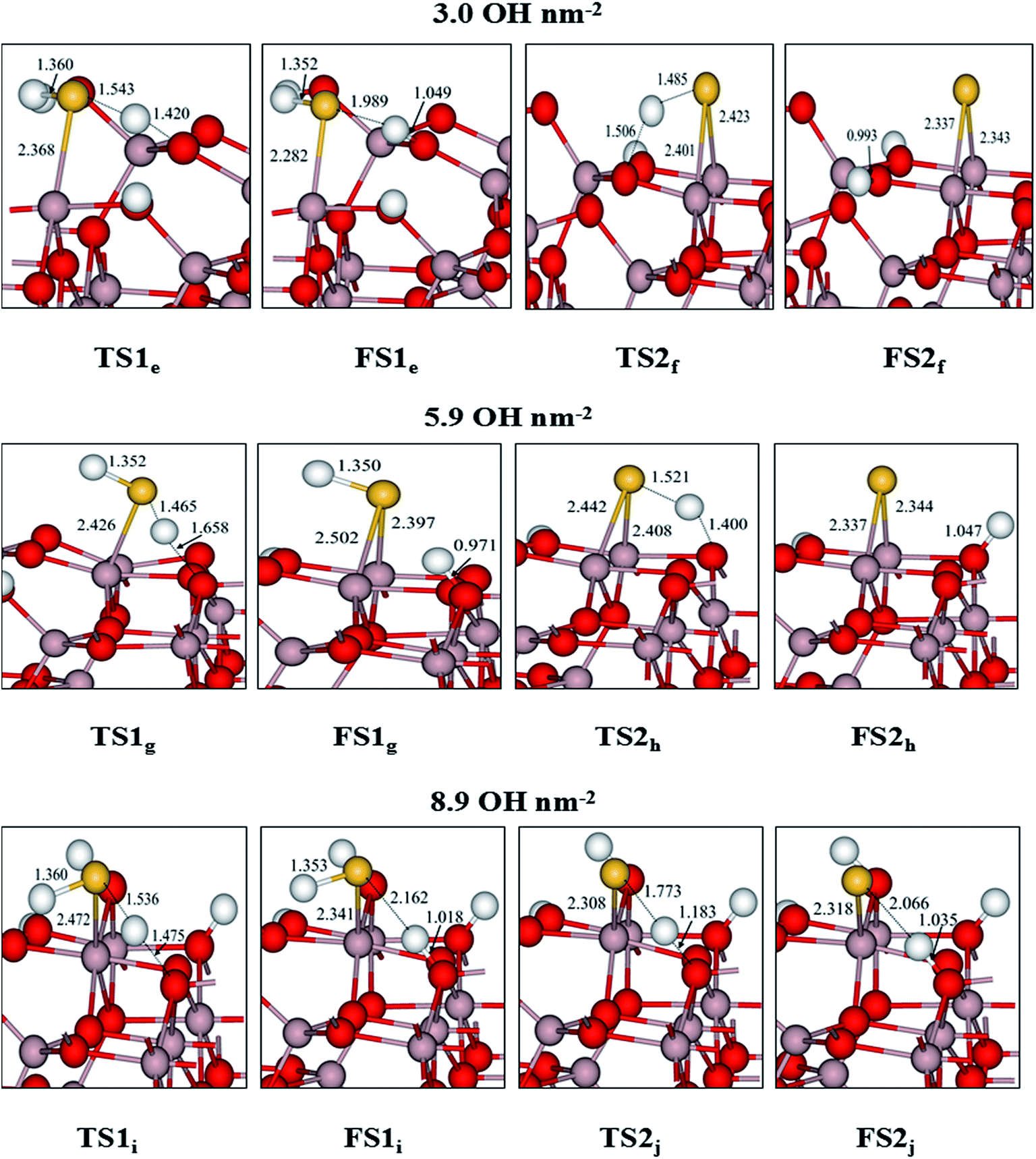 | ||
| Fig. 9 Calculated transition states (TSs) and corresponding final states (FSs) for dissociation of H2S and HS on partially hydrated γ-Al2O3 (110) surfaces. Distances are given in Å (gray, Al; red, O; yellow, S; white, H). | ||
In the second dissociation step, the HS can undergo the dehydrogenation process (HS → H + S) forming H + S(FS2b) by overcoming an activation energy of 51.60 kJ mol−1 in TS2b, as shown in Fig. 6(b). This calculated value of activation energy is as much as 38.38 kJ mol−1 higher than that of the first dissociation step. This result indicates that this dissociation step is slightly difficult from the first dissociation step, and the second step is the rate-determining step in the dissociation reaction. By this reaction, the Al–S and H–S bond distances change from 2.425 and 1.354 Å in the HS to 2.322 and 1.511 Å in the TS2b, finally reaching 2.272 and 1.987 Å in the dissociation state (FS2b). In FS2b, the H and S atoms occupy the O(C) and Al(III) sites, respectively, with a coadsorption energy of −652.14 kJ mol−1.
Further dehydrogenation can take place via TS2d to form surface atomic S and another H atom. In TS2d, the H–S bond is stretched to 1.626 Å from 1.354 Å in HS (see Fig. 4). The activation energy is 77.31 kJ mol−1, which increases by 73.00 kJ mol−1 compared with the parameter of TS1c. After TS2d, the dissociative H atom attaches to the O(C) site, and the S atom diffuses to the Al(II)–Al(III) bridge site from the Al(III) site, forming the structure of FS2d which is similar to the dissociative adsorption geometry of HCN on the γ-Al2O3 (110) surface.63 By comparing the stable adsorption sites of the species between the coadsorption structure (see Fig. 8 FS2d) and the single optimal adsorption structure (see Fig. 4 S(a)), it is found that the most stable coadsorption site is not the superposition of every single optimal adsorption site. The detailed structural parameters are depicted in Fig. 8.
For H2S dissociation on the 3.0 OH per nm2 surface, based on the most stable adsorption structure of H2S (see Fig. 5 (3.0 OH per nm2 surface) H2S) in which the H–S bond is elongated (1.380 Å vs. 1.349 Å in the gas-phase) toward the surface O(C) site, the reaction starts from approaching the H to the O(C) site. The H–S bond distances change from 1.380 Å in the IS to 1.543 Å in the TS1e, finally reaching 1.989 Å in the FS1e. In FS1e, the produced O–H bond is 1.049 Å. The calculated activation energy is 6.73 kJ mol−1, which is similar to that of the dehydrated surface (4.31 kJ mol−1). Meanwhile, the reaction energies show little difference between these two surfaces (−10.03 vs. −20.21 kJ mol−1). As depicted in Fig. 6(a), the activation energies of the first dehydrogenation on the 5.9 and 8.9 OH per nm2 surfaces are 29.19 and 5.23 kJ mol−1, and the corresponding reaction energies are −42.44 and −31.20 kJ mol−1, respectively. It is clear that this step occurs easily, both kinetically and thermodynamically, on these three surfaces.
Immediately following, the second dissociation step was investigated, and the potential energy profile is depicted in Fig. 6(b). The HS (see Fig. 5 (3.0 OH per nm2 surface)) can break the H–S bond to produce FS2f via transition states TS2f with activation energy of 62.81 kJ mol−1. In TS2f, the breaking H–S and forming H–O bonds are 1.485 and 1.506 Å, respectively. After TS2f, this H–O bond length reduces further and reaches 0.993 Å in FS2f. This process is 38.93 kJ mol−1 exothermic. In HS dissociation on the 5.9 OH per nm2 surface, the H atom is abstracted from HS via TS2h to form H + S(FS2h) with an activation energy of 92.06 kJ mol−1 and an exothermicity of 13.22 kJ mol−1. On the 8.9 OH per nm2 surface, the calculated activation energy (TS2j) for H–S bond scission from HS (see Fig. 5 (8.9 OH per nm2 surface)) to produce FS2j is 38.32 kJ mol−1, with a reaction energy of 8.35 kJ mol−1. In TS2j, the H–S bond distance is 1.773 Å, which is stretched 0.407 Å compared with the one of HS (1.366 Å). Detailed structural parameters are shown in Fig. 9.
From the above, it has been shown that the bonding strengths of sulfur-containing species on the dehydrated (110) surface are stronger than those on the dehydrated (100) surface. As the coverage of surface hydroxyl on the (110) surface, the bonding strengths of H2S on these surfaces are ranked in the following order: H2S (adsorbed on D110) > H2S (adsorbed on 3.0 OH per nm2 surface) > H2S (adsorbed on 8.9 OH per nm2 surface) > H2S (adsorbed on 5.9 OH per nm2 surface) > H2S (adsorbed on D100). The adsorption energies of HS on the three hydrated surfaces are nearly equal, and the bonding strengths are in the order: HS (adsorbed on D110) > HS (adsorbed on 5.9 OH per nm2 surface) ≈ HS (adsorbed on 8.9 OH per nm2 surface) ≈ HS (adsorbed on 3.0 OH per nm2 surface) > HS (adsorbed on D100). Comparing the activation energies of the rate-determining step (HS → H + S), it is found that the value on the 5.9 OH per nm2 surface is highest and on the 8.9 OH per nm2 surface is smallest. It is noted that the adsorption energy and dissociative activation energy of H2S have non-linear relationships on these three partially hydrated (110) surfaces. On the 3.0 OH per nm2 surface, the energy level of the Al(II) site is stronger than that of Al(I), therefore, the Al(II) site benefits the adsorption and activation of H2S.45,64 On the 8.9 OH per nm2 and 5.9 OH per nm2 surfaces, the adsorption energy difference of H2S is caused by the hydrogen bonds because of the same adsorption site (Al(I)). The H–S bond lengths of H2S are 1.362 and 1.416 Å on the 8.9 OH per nm2 surface, and the H–S bond lengths of H2S are 1.352 and 1.396 Å on the 5.9 OH per nm2 surface. This shows that the influence of hydrogen bonds on the 8.9 OH per nm2 surface is larger than on the 5.9 OH per nm2 surface. Therefore, the adsorption stability of H2S on the 8.9 OH per nm2 surface is larger than on the 5.9 OH per nm2 surface. The bond lengths of H2S on the 8.9 OH per nm2 surface are longer than on the 5.9 OH per nm2 surface, indicating that H2S dissociation is easier on the 8.9 OH per nm2 surface than on the 5.9 OH per nm2 surface. The result is similar to CH4 dissociation on the hydrated γ-Al2O3 (110) surfaces.44 It seems reasonable that the reaction occurs at 600 K, which is in accordance with the Claus process. The highest activation energy is also only 92.06 kJ mol−1, which is consistent with the result by Bishara et al. (76 kJ mol−1).65 Therefore, H2S can easily dissociate into S species on the involved γ-Al2O3 surfaces.
4. Conclusions
In this paper, the interactions of H2S with γ-Al2O3 surfaces, including partially hydrated (110), and dehydrated (100) and (110) surfaces, were investigated using DFT. Possible adsorption structures and reaction pathways for H2S dissociation were identified. The reported data show that H2S and HS prefer to adsorb on the Al site, and S and H atoms preferentially locate on the Al–O bridge and O sites, respectively. The bonding strengths of these species on the dehydrated (100) surface are weaker than on the dehydrated (110) surface, which is in good agreement with previous experiments. Because of the surface active sites occupied, the bonding strengths of H2S on hydrated (110) surfaces are smaller than on the corresponding dehydrated surfaces.In this dehydrogenation reaction, the activation energy of the second dissociation step becomes higher compared with that of the first step. Therefore, the second step could be the rate-determining step for H2S dissociation on these surfaces. Comparing the dehydrated (110) surfaces, the presence of surface OH groups has an impact on activation energies and reaction energies in H2S dissociation reactions. The 8.9 OH per nm2 surface creates the lowest activation energy for dissociating H2S, with a positive effect on desulfurization, which is in accordance with the Claus reaction occurring at 600 K. Apparently reasonable hydroxyl coverage is beneficial to removal of H2S. Of course, compared with all the activation energies on these involved surfaces, the highest activation energy is merely 92.06 kJ mol−1 on the 5.9 OH per nm2 surface. The results show that H2S decomposition is facile on these involved γ-Al2O3 surfaces, both thermodynamically and kinetically. It was also proven that the γ-Al2O3 desulfurizer is highly efficient for removal of H2S.
Acknowledgements
The authors gratefully acknowledge the financial support of this study by the National Natural Science Foundation of China (21406154), Natural Science Foundation of Shanxi (2013021007-5), and Special/Youth Foundation of Taiyuan University of Technology (2012L041 and 2013T092).References
- X. Li, Y. Wang, Y. Lei and Z. Gu, RSC Adv., 2012, 2, 2302–2307 RSC.
- S. Vallejos, T. Stoycheva, F. E. Annanouch, E. Llobet, P. Umek, E. Figueras, C. Canè, I. Gràcia and C. Blackman, RSC Adv., 2014, 4, 1489–1495 RSC.
- Y. S. Hong, Z. Zhang, Z. Cai, X. Zhao and B. Liu, Energy Fuels, 2014, 28, 6012–6018 CrossRef CAS.
- J. J. Chen, W. W. Li, H. Q. Yu and X. L. Li, AIChE J., 2013, 59, 3824–3833 CrossRef CAS PubMed.
- R. He, F.-F. Xia, J. Wang, C.-L. Pan and C.-R. Fang, J. Hazard. Mater., 2011, 186, 773–778 CrossRef CAS PubMed.
- K. Guo, J. Wen, Y. Zhao, Y. Wang, Z. Zhang, Z. Li and Z. Qian, Environ. Sci. Technol., 2014, 48, 6844–6849 CrossRef CAS PubMed.
- J. Butt, Activation, deactivation, and poisoning of catalysts, Elsevier, 2012 Search PubMed.
- M. Husmann, C. Hochenauer, X. Meng, W. d. Jong and T. Kienberger, Energy Fuels, 2014, 28, 2523–2534 CrossRef CAS.
- X. Ren, L. Chang, F. Li and K. Xie, Fuel, 2010, 89, 883–887 CrossRef CAS PubMed.
- F. Yazdanbakhsh, M. Bläsing, J. A. Sawada, S. Rezaei, M. Müller, S. Baumann and S. M. Kuznicki, Ind. Eng. Chem. Res., 2014, 53, 11734–11739 CrossRef CAS.
- L. Neveux, D. Chiche, J. Perez-Pellitero, L. Favergeon, A.-S. Gay and M. Pijolat, Phys. Chem. Chem. Phys., 2013, 15, 1532–1545 RSC.
- G. Buelna and Y. Lin, Sep. Purif. Technol., 2004, 39, 167–179 CrossRef CAS.
- C. Apesteguia, S. Trevizan, T. Garetto, J. P. de los Reyes and J. Parera, React. Kinet. Catal. Lett., 1982, 20, 1–6 CrossRef CAS.
- B. Guo, L. Chang and K. Xie, Ind. Eng. Chem. Res., 2014, 53, 8874–8880 CrossRef CAS.
- O. Saur, T. Chevreau, J. Lamotte, J. Travert and J.-C. Lavalley, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 427–437 RSC.
- T. Reshetenko, S. Khairulin, Z. Ismagilov and V. Kuznetsov, Int. J. Hydrogen Energy, 2002, 27, 387–394 CrossRef CAS.
- W. C. Content, Petroleum Technology, John Wiley & Sons, Inc, New Jersey, 2007 Search PubMed.
- A. R. Ferreira, M. J. Martins, E. Konstantinova, R. B. Capaz, W. F. Souza, S. S. X. Chiaro and A. A. Leitao, J. Solid State Chem., 2011, 184, 1105–1111 CrossRef CAS PubMed.
- P. S. Prasad, J. W. Bae, S.-H. Kang, Y.-J. Lee and K.-W. Jun, Fuel Process. Technol., 2008, 89, 1281–1286 CrossRef CAS PubMed.
- I. Desyatov, E. Paukshtis and A. Mashkina, React. Kinet. Catal. Lett., 1990, 41, 85–88 CrossRef CAS.
- A. Datta and R. G. Cavell, J. Phys. Chem., 1985, 89, 450–454 CrossRef CAS.
- Y. Okamoto, M. Ohhara, A. Maezawa, T. Imanaka and S. Teranishi, J. Phys. Chem., 1986, 90, 2396–2407 CrossRef CAS.
- R. Glass and R. Ross, J. Phys. Chem., 1973, 77, 2576–2578 CrossRef CAS.
- T. Slager and C. Amberg, Can. J. Chem., 1972, 50, 3416–3423 CrossRef CAS.
- A. Travert, O. Manoilova, A. Tsyganenko, F. Maugé and J. Lavalley, J. Phys. Chem. B, 2002, 106, 1350–1362 CrossRef CAS.
- A. DeRosset, C. Finstrom and C. Adams, J. Catal., 1962, 1, 235–243 CrossRef.
- C. Arrouvel, H. Toulhoat, M. Breysse and P. Raybaud, J. Catal., 2004, 226, 260–272 CrossRef CAS PubMed.
- J. M. Lo, T. Ziegler and P. D. Clark, J. Phys. Chem. C, 2011, 115, 1899–1910 CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Copéret and J. M. Basset, J. Phys. Chem. B, 2006, 110, 23944–23950 CrossRef CAS PubMed.
- P. Hirunsit, K. Faungnawakij, S. Namuangruk and C. Luadthong, Appl. Catal., A, 2013, 460, 99–105 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS PubMed.
- G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef CAS.
- H. Knözinger and P. Ratnasamy, Catal. Rev.: Sci. Eng., 1978, 17, 31–70 Search PubMed.
- G. Paglia, C. Buckley, A. Rohl, B. Hunter, R. Hart, J. Hanna and L. Byrne, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 144110 CrossRef.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2002, 211, 1–5 CrossRef CAS.
- A. Salameh, J. Joubert, A. Baudouin, W. Lukens, F. Delbecq, P. Sautet, J. M. Basset and C. Coperet, Angew. Chem., Int. Ed., 2007, 46, 3870–3873 CrossRef CAS PubMed.
- X. Tan, X. Ren, J. Li and X. Wang, RSC Adv., 2013, 3, 19551–19559 RSC.
- R. Wischert, P. Laurent, C. Copéret, F. o. Delbecq and P. Sautet, J. Am. Chem. Soc., 2012, 134, 14430–14449 CrossRef CAS PubMed.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS PubMed.
- Z. Wang and M. Flytzani-Stephanopoulos, Energy Fuels, 2005, 19, 2089–2097 CrossRef CAS.
- Y. I. Yoon, M. W. Kim, Y. S. Yoon and S. H. Kim, Chem. Eng. Sci., 2003, 58, 2079–2087 CrossRef CAS.
- S. Wilson, J. Solid State Chem., 1979, 30, 247–255 CrossRef CAS.
- X. Krokidis, P. Raybaud, A.-E. Gobichon, B. Rebours, P. Euzen and H. Toulhoat, J. Phys. Chem. B, 2001, 105, 5121–5130 CrossRef CAS.
- S.-F. Peng and J.-J. Ho, J. Phys. Chem. C, 2010, 114, 19489–19495 CAS.
- G. Herzberg, Molecular spectra and molecular structure, vol. 3: Electronic spectra and electronic structure of polyatomic molecules, Van Nostrand Reinhold, New York, 1966, p. 1 Search PubMed.
- K. Huber and G. Herzberg, Constants of diatomic molecules, vol. IV of Molecular spectra and molecular structure, Van Nostrand Reinhold, New York, 1979 Search PubMed.
- Z. Zuo, W. Huang, P. Han, Z. Gao and Z. Li, Appl. Catal., A, 2011, 408, 130–136 CrossRef CAS PubMed.
- J. A. Rodriguez and A. Maiti, J. Phys. Chem. B, 2000, 104, 3630–3638 CrossRef CAS.
- X. Chu, Z. Lu, Y. Zhang and Z. Yang, Int. J. Hydrogen Energy, 2013, 38, 8974–8979 CrossRef CAS PubMed.
- A. Ionescu, A. Allouche, J.-P. Aycard, M. Rajzmann and F. Hutschka, J. Phys. Chem. B, 2002, 106, 9359–9366 CrossRef CAS.
- H.-T. Chen, Y. Choi, M. Liu and M. Lin, J. Phys. Chem. C, 2007, 111, 11117–11122 CAS.
- C. Ren, X. Wang, Y. Miao, L. Yi, X. Jin and Y. Tan, J. Mol. Struct.: THEOCHEM, 2010, 949, 96–100 CrossRef CAS PubMed.
- D. Marrocchelli and B. Yildiz, J. Phys. Chem. C, 2012, 116, 2411–2424 CAS.
- R. Zhang, H. Liu, J. Li, L. Ling and B. Wang, Appl. Surf. Sci., 2012, 258, 9932–9943 CrossRef CAS PubMed.
- M. A. Christiansen, G. Mpourmpakis and D. G. Vlachos, ACS Catal., 2013, 3, 1965–1975 CrossRef CAS.
- W.-F. Huang, H.-T. Chen and M. Lin, J. Phys. Chem. C, 2009, 113, 20411–20420 CAS.
- S. Kim, D. C. Sorescu and J. T. Yates, J. Phys. Chem. C, 2007, 111, 5416–5425 CAS.
- G. Feng, C.-F. Huo, C.-M. Deng, L. Huang, Y.-W. Li, J. Wang and H. Jiao, J. Mol. Catal. A: Chem., 2009, 304, 58–64 CrossRef CAS PubMed.
- A. Bishara, O. Salman, N. Khraishi and A. Marafi, Int. J. Hydrogen Energy, 1987, 12, 679–685 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The differences of adsorption energies and activation energy using different k-point mesh. See DOI: 10.1039/c5ra05443e |
| This journal is © The Royal Society of Chemistry 2015 |