
One-pot synthesis of unsymmetrical squaramides†
Juan V. Alegre-Requena,
Eugenia Marqués-López and
Raquel P. Herrera*
Laboratorio de Organocatálisis Asimétrica, Departamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, E-50009 Zaragoza, Spain. E-mail: raquelph@unizar.es; Tel: +34 976761190
First published on 1st April 2015
Abstract
The results concerning the first one-pot synthesis of unsymmetrical squaramides are reported. This straightforward procedure allows appealing and commonly used squaramide derivatives to be obtained with very good results. The methodology developed in this work saves energy, eliminates the purification steps for intermediate products, reduces costs and leads to better yields compared to those obtained through the traditional “stop-and-go” approach. Moreover, we have proved the efficiency of our process with the synthesis of three biologically active structures, improving the results of the previously reported stepwise syntheses. Interestingly, this simple and cheap method could attract the interest of pharmaceutical and chemical companies aiming to produce these active compounds on a large scale.
Introduction
The development of one-pot syntheses, in which at least two sequential transformations are performed in a single reaction flask, has gained much attention in the last decade,1 especially when these processes can afford chemicals and drug derivatives.1c This growing interest is due to the increasing importance of sustainable chemistry, which involves saving resources and reducing waste. In stepwise syntheses, after each chemical transformation the process is stopped to purify and isolate the reaction intermediates previous to the subsequent reaction pathway. In this context, one-pot approaches could be the best option for reducing time, costs, resources and waste generation at an industrial scale, since these processes eliminate the purification of the intermediates between individual reaction steps, which is, in many cases, where more time and resources are wasted. Moreover, by reducing the number of synthetic steps and removing the intermediates' purification processes, it is possible to reduce the loss of material and, consequently, to increase the overall yield of the reaction sequence.2 Interestingly, from a biological and pharmaceutical point of view, using one-pot reactions is an attractive way to synthesise active compounds through the generation of large compound libraries, due to their ability to generate families of target products easily.New strategies involving squaramides have attracted increasing interest due to their promising properties, becoming a fundamental motif in research.3 These compounds have unique properties that make them appealing for many different areas within chemistry, such as organic synthesis and catalysis and even medicine. In fact, since the pioneering work reported by Rawal and co-workers in 2008![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 using squaramides as excellent hydrogen bond donor catalysts, this area has been experiencing an impressive growth in the organocatalysis field.3a
4 using squaramides as excellent hydrogen bond donor catalysts, this area has been experiencing an impressive growth in the organocatalysis field.3a
Although squaramides have been the focus of many intensive studies,3 to the best of our knowledge, unsymmetrical squaramides have always been synthesised by using multi-step syntheses, which require purifying the reaction intermediates or changing the reaction media between synthetic steps. Therefore, the novel one-pot approach for synthesizing squaramides described in this work requires less time and resources and is cleaner than previously reported syntheses (Scheme 1).
 | ||
| Scheme 1 One-pot reaction of squaramides. | ||
This work was intended to develop a more straightforward method for synthesizing squaramides that would enable this field to progress and evolve more quickly without the disadvantages of previous standard approaches. Moreover, we performed these syntheses without an excess of reactants, which proves these to be atom-economic processes.5 This not only saves resources, but generally also leads to obtaining pure final products after a simple filtration.
In this work, we report our appealing results concerning the first one-pot synthesis of a great number of squaramides (Scheme 1).6 Furthermore, in order to demonstrate the wide application of this protocol, the syntheses of three biologically active structures have been successfully accomplished (Fig. 1).
 | ||
| Fig. 1 Target molecules synthesised. | ||
Results and discussion
Focusing on the first step of the one-pot process and in order to check the viability of our idea, we performed an initial screening of solvents and concentration of the model reaction, shown in Table 1. We analysed which solvent presented the highest formation of product 3a, which is the first product obtained in the equivalent “stop-and-go” syntheses. Consequently, 3a is the first expected intermediate formed in the current one-pot approach and its formation is the limiting step of this reaction. The first amine addition is crucial in this method because an incomplete conversion of the initial reagents leads to lower yields of final squaramides 5. This decrease in the yield occurs because a smaller amount of intermediate 3 is available for the second amine addition, and the remaining squarate 1 could also react with the second amine. Thus, the generated amount of intermediate 3 limits the overall yield of the final squaramides in this one-pot procedure (Scheme 1).
|
|||
|---|---|---|---|
| Entry | Solvent | Time (h) | Yielda (%) |
| a Conversion calculated by 1H-NMR using dimethyl fumarate as the internal standard.b No reaction.c Reaction performed with 0.25 mL of MeOH. | |||
| 1 | CHCl3 | 24 | 47 |
| 2 | MeOH | 24 | 59 |
| 3 | EtOH | 24 | 53 |
| 4 | CH3CN | 24 | 46 |
| 5 | DMSO | 24 | n.r.b |
| 6 | DMF | 24 | n.r.b |
| 7 | 1,4-Dioxane | 24 | n.r.b |
| 8 | Toluene | 24 | n.r.b |
| 9c | MeOH | 24 | 69 |
| 10c | MeOH | 85 | >95 |

Among all the studied solvents, we found MeOH as the best choice for the synthesis of intermediate 3a (entry 2), whereas no reaction was observed with some polar solvents such as DMSO, DMF or 1,4-dioxane (entries 5–7). Non-polar solvents such as toluene also showed no reaction (entry 8). This was also observed by Luk and coworkers, who demonstrated that the addition of the second amine in squaramide synthesis was faster when hydrogen-bond donors were employed, concluding that solvents with a higher hydrogen-donor character led to better reactions rates.3e In our work, this conclusion can be also stated since the reaction rates observed in the additions of the first amines depended on the hydrogen-bond donor acidities7 of the solvents and not on their dielectric constants. However, the use of pure water as the solvent was not effective since the formation of the final squaramides was low due to the high insolubility of intermediate 3a in this medium. Moreover, the hydrolysis of squarate 1a in water produced different byproducts as observed previously in other works.8 Increasing the concentration of the reaction had a pronounced positive effect on the yield. When using 0.25 mL of MeOH, 3a was obtained in 69% yield, compared to the 59% yield obtained when 0.5 mL of MeOH was used (entry 9 vs. entry 2). Additionally, testing the yield of the reaction at different times revealed that conversion of 3a was >95% after 85 h (entry 10). This is of a great importance since MeOH has been considered one of the preferred green solvents.7
With these values in hand, we also explored the viability of the formation of intermediates 3b, c, f when using three other common anilines (2b, c, f), in order to prove if MeOH was also a useful solvent for other reagents (Scheme 2).
 | ||
| Scheme 2 Test of different anilines. Conversion calculated by 1H-NMR using dimethyl fumarate as the internal standard. | ||
The screening of different anilines revealed that the process was also applicable to other substrates with excellent yields for the synthesis of intermediate squaramide monoamines 3b, c, f (Scheme 2).
After confirming the compatibility of this method with different anilines, the viability of the one-pot process was tested by incorporating the third reaction component (amine 4) in the reaction medium after the formation of intermediate 3. A great number of anilines and amines were tested for the efficient formation of structures 5, which we are currently researching as promising organocatalysts for asymmetric catalysis (Table 2). Among them there were 15 new squaramide structures (entries 1–16) and 11 structures that had previously been reported (entries 17–29). In our one-pot procedure, first we added amine 2a–i in one portion into a solution of squarate 1a in MeOH, and after a reaction time of t1, we added the second amine 4a–l dissolved in MeOH. After a time of t2, the reaction was stopped and purified. Generally, the final products were purified by filtration, although in a few cases, these products were purified by column chromatography.

| Entry | NH2R1 (2a–i)/MeOH (mL) | NH2R2 (4a–l)/MeOH (mL) | t1 (h) | t2 (h) | Product | Yieldb (%) |
|---|---|---|---|---|---|---|
| a Experimental conditions: to a mixture of squarate 1a (0.2 mmol) in MeOH (0.25–1 mL), amine 2a–i (0.2 mmol) was added in one portion at room temperature. After the reaction time t1, amine 4a–l (0.2 mmol) was further added dissolved in MeOH (1–4.5 mL). After t2, adduct 5 was filtrated under vacuum, placed at −22 °C for 30 minutes and the solid was washed with cold MeOH (1 mL).b Isolated yield. The results in parenthesis are the yields obtained by previous authors in the respective two-step syntheses.c Reaction performed for 1 mmol of reagents.d Reaction performed for 0.15 mmol of reagents.e Purified by column chromatography (SiO2 using Hex:EtOAc 6:4 to EtOAc:MeOH 9:1).f Purified by column chromatography (SiO2 using EtOAc to EtOAc:MeOH 7:3).g Reaction performed for 0.4 mmol of reagents.h Purified by column chromatography (SiO2 treated with Et3N, using CH2Cl2 to CH2Cl2:MeOH 98:2).i Purified by column chromatography (SiO2, using Hex:EtOAc 6:4 to EtOAc:MeOH 7:3). |
||||||
| 1 | 2a/0.25 | 4a/1.75 | 80 | 20 | 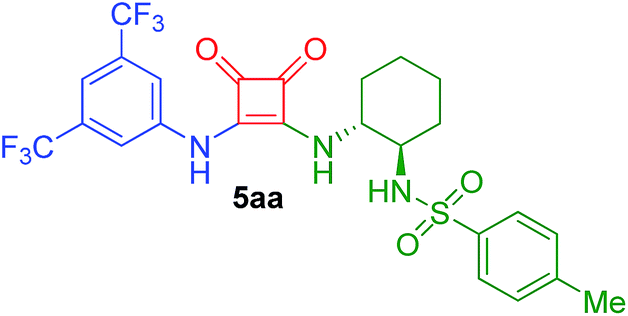 |
75 |
| 2 | 2b/0.5 | 4a/1.5 | 26 | 25 |  |
82 |
| 3 | 2d/1 | 4a/1 | 2 | 2 |  |
72 |
| 4c | 2d/5 | 4a/5 | 2.5 | 4 | 80 | |
| 5 | 2e/0.5 | 4a/1.5 | 3 | 7 |  |
85 |
| 6 | 2g/0.5 | 4a/1.5 | 20 | 3 |  |
72 |
| 7 | 2a/0.25 | 4b/1.75 | 82 | 10 |  |
77 |
| 8 | 2b/0.5 | 4b/1.5 | 48 | 14 |  |
>95 |
| 9c | 2a/1.25 | 4c/5.75 | 82 | 3 |  |
84 |
| 10 | 2a/0.25 | 4d/1.75 | 82 | 14 |  |
60 |
| 11 | 2a/0.25 | 4e/1.75 | 49 | 3 |  |
61 |
| 12 | 2b/0.5 | 4e/1.5 | 35 | 5 | 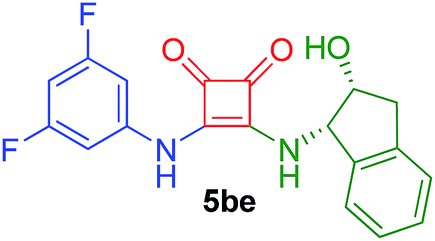 |
88 |
| 13 | 2b/0.5 | 4f/4.5 | 30 | 3 |  |
82 |
| 14 | 2f/0.75 | 4g/1.25 | 66 | 3 | 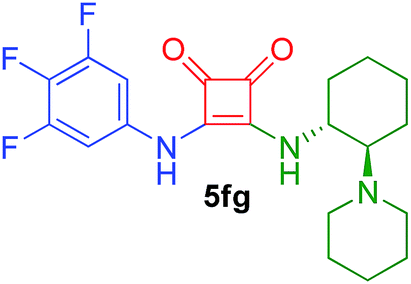 |
82 |
| 15d | 2h/0.75 | 4g/1.25 | 65 | 23 |  |
68e |
| 16d | 2h/0.75 | 4h/1.25 | 67 | 25 | 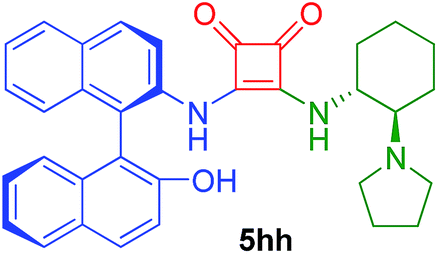 |
50f |
| 17g | 2a/0.5 | 4g/3.5 | 77 | 2.5 | 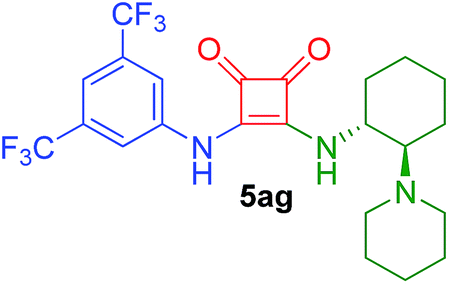 |
80h (43)10 (65)9 |
| 18g | 2c/1 | 4g/3 | 48 | 3 |  |
77 (58)10 (62)11 |
| 19 | 2a/0.25 | 4j/1.75 | 72 | 48 |  |
45i |
| 20g | 2a/0.5 | 4j/3.5 | 76 | 45 | 52i (49)12 | |
| 21 | 2c/0.5 | 4j/1.5 | 48 | 48 |  |
48 or 64i (74)12 |
| 22g | 2g/1 | 4j/3 | 26 | 48 | 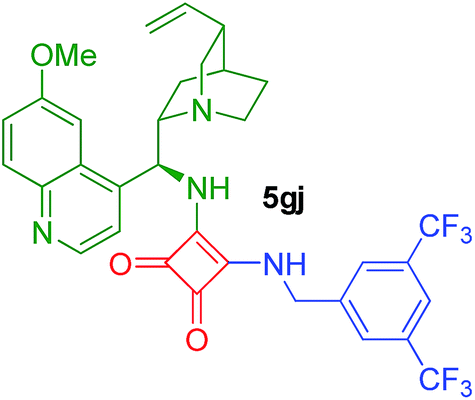 |
56 or 74i (78)13 |
| 23 | 2a/0.25 | 4k/1.75 | 77 | 25 |  |
67i (49)12 |
| 24 | 2c/0.5 | 4k/1.5 | 48 | 48 |  |
68 or 78i (72)12 |
| 25 | 2g/0.5 | 4l/1.5 | 22 | 48 |  |
43 |
| 26g | 2g/1 | 4l/3 | 26 | 48 | 53 or 67i (61)4 | |
| 27 | 2g/0.5 | 4g/1.5 | 21 | 3 |  |
43 (34)14 |
| 28 | 2i/1 | 4g/1 | 48 | 23 | 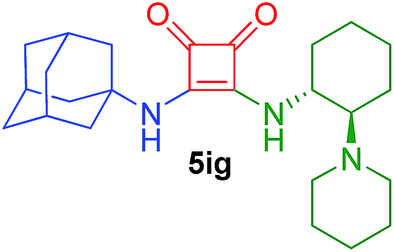 |
53 (30)15 |
| 29 | 2a/0.25 | 4i/1.75 | 74 | 3 |  |
60 (29)10 (48)9 |
In every reaction, final squaramides 5 were produced in moderate to very good yields (from 43% to >95% yield). Unless otherwise indicated, the products were purified by a simple filtration of the final solid under vacuum conditions and washed with cold MeOH. We were encouraged to observe that it was possible to slightly increase the yield of the process by performing the reactions on a larger scale (with 0.4 or 1 mmol of reagents, entries 4, 20 and 26). When performing these reactions at a larger scale, the loss of material reduced the overall yield less than at a small scale (72% vs. 80% in entries 3 and 4, 45% vs. 52% in entries 19 and 20, and 43% vs. 53% in entries 25 and 26). This is also interesting from an industrial point of view because even better yields could be expected at an industrial scale. The concentration of each reaction has been studied and optimised individually, based on the different insolubilities of the reagents and the final products. However, despite each reaction having its own optimization process, only slight modifications in the amounts of MeOH used in the reactions were necessary (Table 1). In most of the comparisons, the new one-pot procedure gave better results than the previous two-step approaches, showing an increase in yield of up to 23% compared to the highest values previously reported (entry 28). However, squaramides 5cj (entry 21) and 5gj (entry 22) were obtained in slightly lower yields than those previously reported in the “stop-and-go” procedures. In our case, the slightly lower yield could be attributed to the small scale at which the reactions were performed, but better results could be expected when these processes are scaled-up as we have already demonstrated for some examples.
Interestingly, the commercially available squaramide organocatalyst 5cg was obtained with a better yield when compared to the previously reported syntheses of the same compound (see Scheme 3 and Table 2, entry 18).10,11 In the previous procedures, the authors used an excess of amine 4g (1.5 equiv.), the most expensive part of this squaramide structure, in the second reaction step. However, we performed the reaction using equimolar amounts of each reagent in order to reduce the waste and costs involved in the process. Moreover, we stopped the reaction 3 hours after adding second amine 4g, which is a notably shorter reaction time than those in previous procedures (24 h, Scheme 3). As mentioned above, the yield of this process may be increased if the reaction is scaled up.
 | ||
| Scheme 3 Syntheses of commercially available squaramide 5cg. | ||
After these promising results, we considered extending the applicability of our simple, cheap and straightforward methodology to the synthesis of biologically active compounds, in order to enlarge the scope of this method and to demonstrate the importance and applications of this pioneering one-pot procedure.
Recently, Moreno, Costa and co-workers have discovered an effective antiparasitic agent that acts as an anti-Chagasic drug (Scheme 4). Squaramide 10 has resulted to be a promising candidate in the preliminary in vivo studies on acute and chronic phases of Chagas disease and has shown low toxicity.16 This disease is a tropical parasitic infection that mainly affects to the poorest rural areas of Latin America, and although there are treatments for this illness, more efforts in research as well as more efficient and less expensive drugs are required to combat Chagas disease.17 In this context, squaramide 10, as a low-cost drug, could represent an interesting alternative to the medicines that are currently being used for this disease, the later having severe side effects and not being effective for chronic patients.
 | ||
| Scheme 4 Syntheses of anti-Chagasic squaramide 10. | ||
The results previously reported for the production of this interesting compound16 were improved when using our synthetic method. In the previous synthesis, the authors needed three sequential transformations with an overall yield for the last two steps of around 67.5%. In contrast, we started our synthesis from squarate 1a dissolved in MeOH (0.25 mL), and then added amine 7. In the same vessel, after 2.5 h we added BuNH2 (9) dissolved in MeOH (1.75 mL). After 12.5 h, the product was then purified by column chromatography, obtaining the final product 10 in a 78% yield. Therefore, the yield achieved using our one-pot procedure was higher than that of the previous three-step process.
This simple and inexpensive method might attract the interest of pharmaceutical companies for the production of this compound at a larger scale. This could help to overcome one of the drawbacks that this disease now presents: the high cost of the drugs used to fight it, which makes the treatment inaccessible for many people affected by this illness.
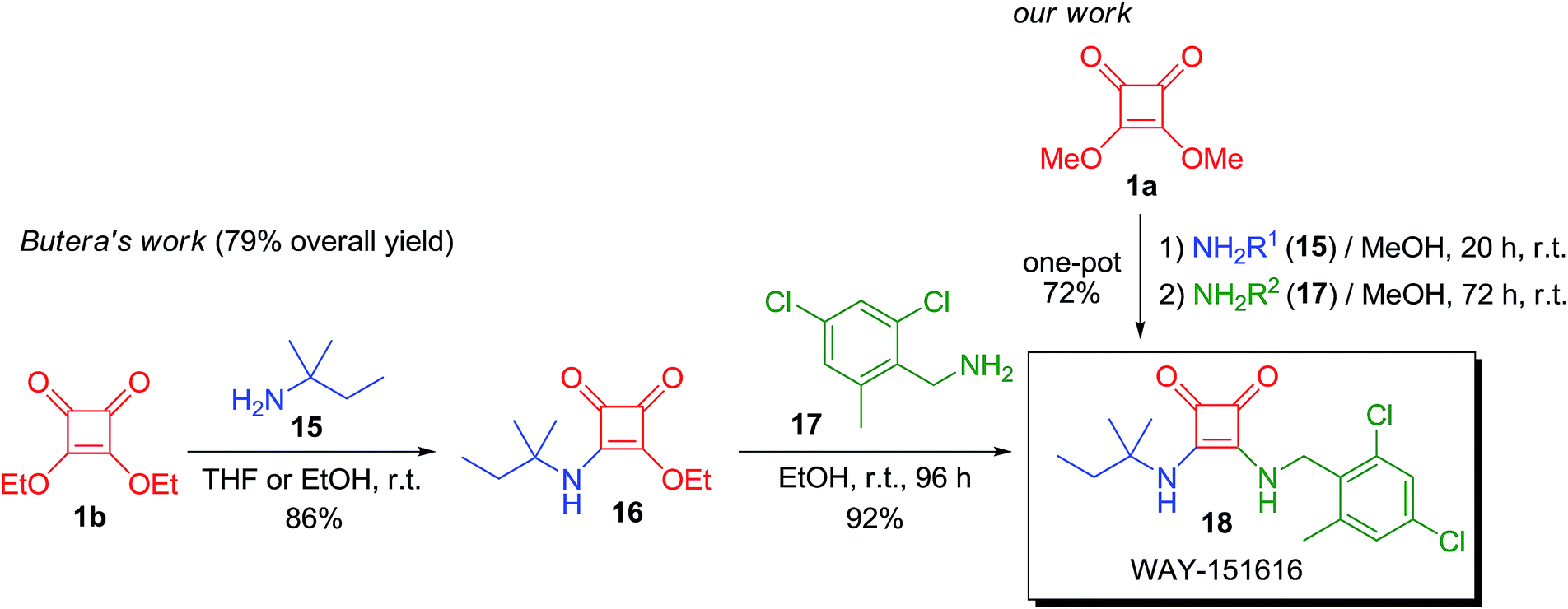
We also prepared two other active compounds, 14 and 18, as potent smooth muscle relaxants (Schemes 5 and 6). These two bladder-selective KATP-channel openers18 exhibit remarkable oral efficacy in a rat hypertrophied bladder model of urge urinary incontinency,19 a disease that affects between 10 to 20 percent of the worldwide population and causes enormous health care expenses. The therapeutic drugs that are currently being used to treat this disease, such as tolterodine, a muscarinic antagonist, present some drawbacks that limit their use and special care must be taken when supplying these drugs. These side effects have made it necessary to search for new candidates with different mechanisms of action that show enhanced efficacy and work in a wide range of patients. In this sense, WAY-133537 (14) and WAY-151616 (18) have been revealed as promising drug candidates since they show bladder selectivity without causing severe cardiovascular side effects.
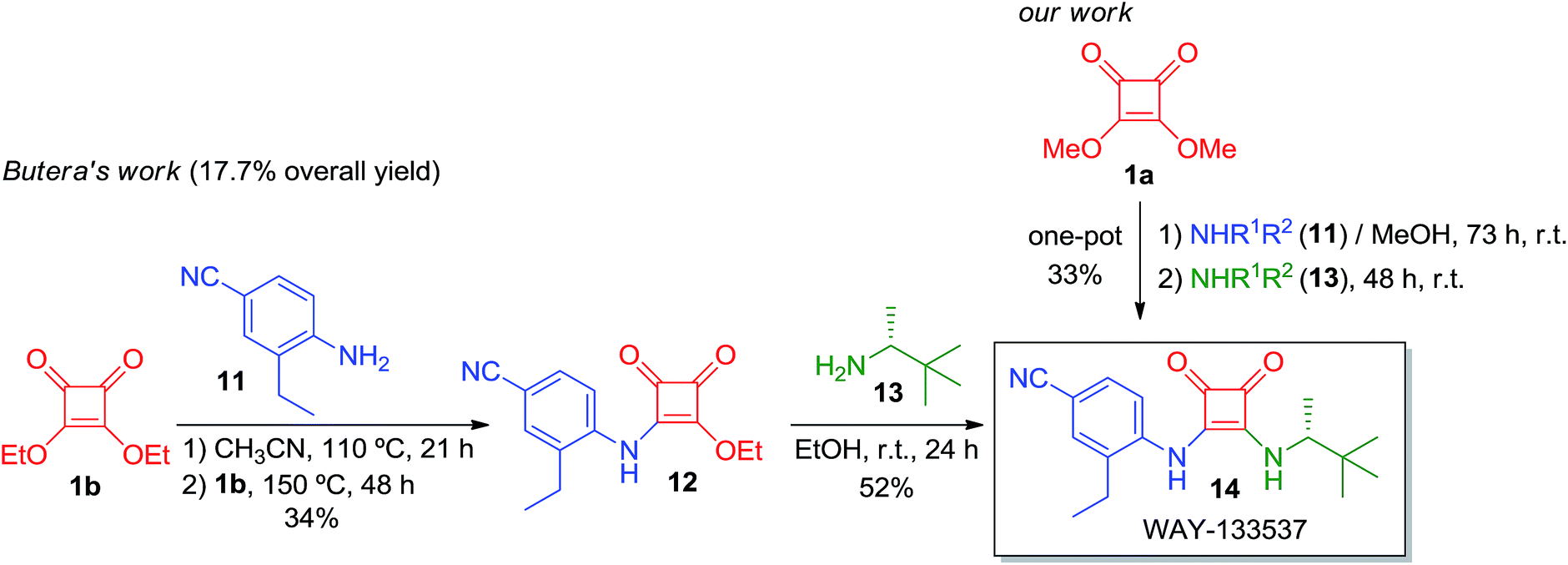 | ||
| Scheme 5 Syntheses of biologically active WAY-133537 (14). | ||
 | ||
| Scheme 6 Syntheses of biologically active WAY-151616 (18). | ||
The pioneering synthesis of compound 14 is described in Scheme 5 with an overall yield of 17.7% (scale of 5.88 mmol).20 Squaramide derivative 14 has been successfully tested in urinary incontinence disorder, but it is also efficient in the treatment of other diseases related to smooth muscle contraction such as hypertension, asthma, premature labor, irritable bowel syndrome, congestive heart failure, angina and cerebral vascular disease.21 The one-pot method that we have developed was also successfully employed in the synthesis of this appealing drug and it was possible to synthesise it in a 33% yield (at a scale of 0.2 mmol). In this case, the initial amount of solvent was slightly modified due to the high insolubility of intermediate 12, starting from squarate 1a and amine 11 dissolved in 1 mL of MeOH. After 73 h of reaction, we added amine 13 as a liquid without MeOH. Finally, after 2 days the precipitate was filtered under vacuum and the solid was washed with cold MeOH (1 mL), obtaining the final product 14 in a 33% yield.
In addition, we also prepared WAY-151616 (18) using our new synthetic approach. Squaramide 18 is another potent potassium channel opener with specificity towards bladder tissue, which has also shown promising results as potential drug in the treatment of urge urinary incontinence and it is already being produced industrially (Scheme 6).21d,22
Biologically active squaramide 18 was prepared by Butera and co-workers following the procedure described in Scheme 6, with an overall yield of 79%.23 Employing our one-pot method, we reached a 72% yield in one single step. It is noteworthy that the authors performed this synthesis to obtain around 28 g of compound 18 while we were operating on a very small scale (0.4 mmol of product, 136 mg). Therefore, a higher yield should be expected when employing our methodology on a larger scale. When we performed the reaction at a smaller scale (0.2 mmol), the final product was rendered with a 62% yield, displaying the importance of scale and the possibility of improving our method by increasing the amount of product synthesised.
Conclusions
In summary, we have reported the first one-pot synthesis of squaramides with results ranging from moderate to very good. This is a simpler procedure for obtaining appealing squaramide derivatives when compared to the previously developed “stop-and-go” processes. This method is energy-saving, avoids time consuming purification steps and reduces costs, leading, in most cases, to better yields when compared to those obtained through the traditional sequential approach.Furthermore, we have proved the utility of our process with the synthesis of three biologically active structures, improving upon the results of the previous stepwise syntheses. Moreover, we have been able to synthesise the commercially available squaramide 5cg, which is being employed extensively as an organocatalyst, using this one-pot method and obtaining higher yields than those of previous works. This simple and inexpensive method could attract the interest of pharmaceutical and chemical companies aiming to produce these compounds at a large scale. We believe that our work could be a pivotal and crucial precedent in the field of squaramides due to the simplicity of the operational procedure reported here.
Experimental
General Experimental methods
When needed, purification of reaction products was carried out by flash chromatography using silica-gel (0.063–0.200 mm). Analytical thin layer chromatography was performed on 0.25 mm silica gel 60-F plates. ESI ionization method and mass analyzer type MicroTof-Q were used for the ESI measurements. 1H NMR spectra were recorded at 400 and 300 MHz; 13C APT-NMR spectra were recorded at 100 and 75 MHz; DMSO-d6 and CDCl3 as the solvents. Chemical shifts were reported in the δ scale relative to residual CHCl3 (7.26 ppm) and DMSO (2.50 ppm) for 1H NMR and to the central line of CHCl3 (77 ppm) and DMSO (39.43 ppm) for 13C APT-NMR.All commercially available solvents and reagents were used as received. The 1H and 13C NMR spectra for compounds 5ag,9 5cg,10 5aj,12 5cj,12 5gj,13 5ak,12 5ck,12 5gl,4 5gg,14 5ig,15 5ai,9 10,16 14,20 and 1823 are consistent with values previously reported in the literature.
Representative procedure for the one-pot synthesis of squaramides 5
To a mixture of 3,4-dimethoxy-3-cyclobutene-1,2-dione (1a) (0.2 mmol) in MeOH (0.25–1 mL), amine 2a–i was firstly added in one portion at room temperature. After the corresponding reaction time (t1) (see Table 1), amine 4a–l (1 equivalent) was then added dissolved in MeOH (1.75–1 mL). After the corresponding reaction time (t2) (see Table 1), the product was purified by filtration or by column chromatography. Yields are reported in Table 1 and pure compounds were obtained as stable solids.One-pot synthesis of antiparasitic agent 10
To a mixture of 3,4-dimethoxy-3-cyclobutene-1,2-dione (1a) (0.2 mmol) in MeOH (0.25 mL), 0.2 mmol of N,N,N′-trimethyl-1,3-propanediamine (7) was firstly added at room temperature in one portion. After 2.5 h, n-butylamine (9) (0.2 mmol) was dissolved in MeOH (1.75 mL) and added to the reaction mixture in one portion. After 12.5 h, the solvent was evaporated in vacuum and the product was purified by column chromatography (SiO2, EtOAc:MeOH 1:1 to EtOAc:MeOH:Et3N 5:5:0.1). Final anti-Chagasic drug 10 was obtained as a white solid in 78% yield.
One-pot synthesis of WAY-133537 (14)
To a mixture of 3,4-dimethoxy-3-cyclobutene-1,2-dione (1a) (0.2 mmol) in MeOH (1 mL), 0.2 mmol of 4-amino-3-ethylbenzonitrile (11) was firstly added at room temperature in one portion. After 73 h, 0.2 mmol of (R)-3,3-dimethyl-2-butylamine (13) was added to the reaction mixture in one portion. After 48 h, the test tube was placed in a freezer (−22 °C) for 30 minutes to facilitate the overall precipitation of final drug 14. The product was filtered under vacuum and washed with cold MeOH (1 mL). Final bladder-selective agonist KATP channel 14 was obtained as a green solid in 33% yield.One-pot synthesis of WAY-151616 (18)
To a mixture of 3,4-dimethoxy-3-cyclobutene-1,2-dione (1a) (0.4 mmol) in MeOH (0.5 mL), 0.4 mmol of 1,1-dimethylpropylamine (15) were firstly added at room temperature in one portion. After 20 h, 0.4 mmol of 2,4-dichloro-6-methylbenzylamine (17) were dissolved in MeOH (3.5 mL) and added to the reaction mixture in one portion. After 72 h, the solvent was evaporated under vacuum and the product was purified by column chromatography (SiO2, hexane/EtOAc 6:4 to 0:1 and EtOAc/MeOH 9:1). Final bladder-selective agonist KATP channel 18 was obtained as a white solid in 72% yield.
N-((1R,2R)-2-(2-(3,5-Bis(trifluoromethyl)phenylamino)-3,4-dioxocyclobut-1-enylamino)cyclohexyl)-4-methylbenzenesulfonamide (5aa)
Following the general procedure, compound 5aa was obtained after 100 h of reaction at room temperature as a white solid in 75% yield. [α]25D = +34 (c 0.55, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 10.02 (br s, 1H), 8.06 (s, 2H), 7.74 (br d, J = 8.5 Hz, 1H), 7.69 (s, 1H), 7.63 (br d, J = 8.9 Hz, 1H), 7.59 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 3.62–3.52 (m, 1H), 3.18–3.05 (m, 1H), 2.13 (s, 3H), 1.92–1.89 (m, 1H), 1.72–1.37 (m, 4H), 1.35–1.10 (m, 3H). 13C APT-NMR (100 MHz, DMSO-d6) δ 183.8 (1C), 179.8 (1C), 169.5 (1C), 162.0 (1C), 141.7 (1C), 141.1 (1C), 139.3 (1C), 131.4 (q, J = 34.2 Hz, 2C), 130.9 (2C), 125.9 (2C), 123.1 (q, J = 271.7 Hz, 2C), 117.6 (2C), 114 (1C), 57.3 (1C), 56.8 (1C), 32.7 (1C), 32.6 (1C), 24.0 (1C), 24.0 (1C), 20.5 (1C). IR (KBr film) (cm−1) ν 3284, 3177, 2924, 2854, 1793, 1659, 1582, 1559, 1457, 1379, 1277, 1191, 1163, 1151, 1129, 1093, 880, 754, 666. HRMS (ESI+) calcd C25H24F6N3O4S 576.1392; found 576.1360 [M + H].N-((1R,2R)-2-(2-(3,5-Difluorophenylamino)-3,4-dioxocyclobut-1-enylamino)cyclohexyl)-4-methylbenzenesulfonamide (5ba)
Following the general procedure, compound 5ba was obtained after 51 h of reaction at room temperature as a white solid in 82% yield. [α]25D = +42 (c 0.49, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 9.69 (br s, 1H), 7.70 (d, J = 8.5 Hz, 1H), 7.67 (d, J = 8.8 Hz, 1H), 7.59 (d, J = 7.9 Hz, 2H), 7.23–7.18 (m, 4H), 6.86 (t, J = 9.2 Hz, 1H), 3.60–3.53 (m, 1H), 3.16–3.09 (m, 1H), 2.17 (s, 3H), 1.92–1.89 (m, 1H), 1.64–1.11 (m, 7H). 13C APT-NMR (100 MHz, DMSO-d6) δ 183.6 (1C), 179.6 (1C), 169.2 (1C), 163.0 (d, J = 242.7 Hz, 1C), 162.9 (d, J = 243.9 Hz, 1C), 162.4 (1C), 141.9 (1C), 141.8 (t, J = 13.8 Hz, 1C), 139.3 (1C), 129.2 (2C), 125.9 (2C), 100.9 (d, J = 29.6 Hz, 2C), 97.1 (t, J = 26.6 Hz, 1C), 57.2 (1C), 56.9 (1C), 32.8 (1C), 32.6 (1C), 24.0 (1C), 24.0 (1C), 20.6 (1C). IR (KBr film) (cm−1) ν 3369, 3289, 3189, 3044, 2924, 2854, 1797, 1664, 1616, 1575, 1536, 1461, 1377, 1319, 1205, 1158, 1118, 1089, 994, 854, 749, 670, 571. MS (ESI+) 476.2 [M + H].N-((1R,2R)-2-(2-(4-tert-Butylphenylamino)-3,4-dioxocyclobut-1-enylamino)cyclohexyl)-4-methylbenzenesulfonamide (5da)
Following the general procedure, compound 5da was obtained after 6.5 h of reaction at room temperature as a white solid in 80% yield. [α]24D = +48 (c 0.52, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 9.40 (br s, 1H), 7.65 (br d, J = 8.1 Hz, 1H), 7.59 (d, J = 8.2 Hz, 2H), 7.51 (br d, J = 7.1 Hz, 1H), 7.43–7.35 (m, 4H), 7.21 (d, J = 8.1 Hz, 2H), 3.60–3.43 (m, 1H), 3.12–3.07 (m, 1H), 2.13 (s, 3H), 1.94–1.87 (m, 1H), 1.69–1.11 (m, 7H), 1.28 (s, 9H). 13C APT-NMR (100 MHz, DMSO-d6) δ 183.0 (1C), 179.8 (1C), 168.6 (1C), 163.5 (1C), 144.9 (1C), 142.0 (1C), 139.2 (1C), 136.5 (1C), 129.2 (2C), 125.9 (4C), 117.5 (2C), 57.0 (1C), 56.9 (1C), 33.9 (1C), 32.9 (1C), 32.7 (1C), 31.1 (3C), 24.1 (1C), 24.0 (1C), 20.6 (1C). IR (KBr film) (cm−1) ν 3343, 3195, 2924, 2854, 1795, 1660, 1604, 1568, 1535, 1460, 1375, 1322, 1268, 1162, 1143, 1094, 829, 723, 667. HRMS (ESI+) calcd C27H34N3O4S 469.2270; found 469.2233 [M + H].N-((1R,2R)-2-(2-(4-Methoxyphenylamino)-3,4-dioxocyclobut-1-enylamino)cyclohexyl)-4-methylbenzenesulfonamide (5ea)
Following the general procedure, compound 5ea was obtained after 10 h of reaction at room temperature as a white solid in 85% yield. [α]24D = +40 (c 0.58, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 9.35 (br s, 1H), 7.64 (br d, J = 8.4 Hz, 1H), 7.59 (d, J = 8.2 Hz, 2H), 7.42 (br d, J = 7.1 Hz, 1H), 7.37 (d, J = 8.7 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 9.0 Hz, 2H), 3.74 (s, 3H), 3.59–3.50 (m, 1H), 3.16–3.06 (m, 1H), 2.16 (s, 3H), 1.91–1.88 (m, 1H), 1.67–1.17 (m, 7H). 13C APT-NMR (100 MHz, DMSO-d6) δ 182.6 (1C), 179.9 (1C), 168.3 (1C), 163.4 (1C), 155.0 (1C), 142.0 (1C), 139.2 (1C), 132.2 (1C), 129.2 (2C), 125.9 (2C), 119.2 (2C), 114.5 (2C), 56.9 (1C), 56.9 (1C), 55.2 (1C), 32.9 (1C), 32.8 (1C), 24.1 (1C), 24.0 (1C), 20.7 (1C). IR (KBr film) (cm−1) ν 3348, 3266, 2922, 2853, 1790, 1646, 1613, 1564, 1538, 1515, 1477, 1368, 1323, 1251, 1160, 1094, 1077, 829, 810, 665. HRMS (ESI+) calcd C24H28N3O5S 470.1750; found 470.1729 [M + H].N-((1R,2R)-2-(2-(3,5-Bis(trifluoromethyl)benzylamino)-3,4-dioxocyclobut-1-enylamino)cyclohexyl)-4-methylbenzenesulfonamide (5ga)
Following the general procedure, compound 5ga was obtained after 23 h of reaction at room temperature as a white solid in 72% yield (85 mg). [α]24D = +33 (c 0.49, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 8.09 (s, 2H), 8.05 (s, 1H), 7.80 (br s, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.57 (br s, 1H), 7.38 (br s, 1H), 7.27 (d, J = 8.1 Hz, 2H), 4.90 (br s, 2H), 3.60–3.45 (m, 1H), 3.04–2.94 (m, 1H), 2.33 (s, 3H), 1.86–1.80 (m, 1H), 1.60–1.01 (m, 7H). 13C APT-NMR (100 MHz, DMSO-d6) δ 182.1 (1C), 168.0 (1C), 142.5 (1C), 141.1 (2C), 139.3 (2C), 130.4 (q, J = 32.5 Hz, 2C), 129.3 (2C), 128.4 (2C), 126.0 (2C), 123.2 (q, J = 271.2 Hz, 2C), 121.1 (1C), 56.9 (1C), 56.7 (1C), 45.6 (1C), 32.8 (1C), 32.1 (1C), 24.0 (2C), 20.8 (1C). IR (KBr film) (cm−1) ν 3373, 3166, 2924, 2854, 1798, 1650, 1569, 1496, 1456, 1377, 1321, 1268, 1237, 1203, 1186, 1171, 1095, 894, 729, 706, 682, 568. MS (ESI+) 590.2 [M + H].(S)-3-(3,5-Bis(trifluoromethyl)phenylamino)-4-(2-hydroxy-1,2,2-triphenylethylamino)cyclobut-3-ene-1,2-dione (5ab)
Following the general procedure, compound 5ab was obtained after 92 h of reaction at room temperature as a white solid in 77% yield. [α]25D = −97 (c 0.40, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 10.40 (br s, 1H), 8.74 (br d, J = 9.6 Hz, 1H), 8.00 (s, 2H), 7.67 (d, J = 7.5 Hz, 2H), 7.63 (s, 1H), 7.41–7.05 (m, 13H), 6.71 (br s, 1H), 6.22 (br d, J = 9.7 Hz, 1H). 13C APT-NMR (100 MHz, DMSO-d6) δ 184.6 (1C), 179.9 (1C), 168.3 (1C), 162.5 (1C), 145.5 (1C), 144.1 (1C), 140.8 (1C), 138.9 (1C), 131.2 (q, J = 31.6 Hz, 2C), 128.8 (2C), 128.0 (2C), 127.5 (1C) 127.4 (2C), 127.0 (1C), 126.7 (1C), 126.5 (1C), 126.3 (1C), 126.1 (2C), 126.0 (2C), 123.0 (q, J = 271.0 Hz, 2C), 118.0 (2C), 114.6 (1C), 80.4 (1C), 63.3 (1C). IR (KBr film) (cm−1) ν 3506, 3220, 3135, 2923, 2854, 1804, 1660, 1604, 1575, 1546, 1506, 1469, 1380, 1280, 1184, 1128, 1062, 889, 752, 699. HRMS (ESI+) calcd C32H23F6N2O3 597.1613; found 597.1599 [M + H].(S)-3-(3,5-Difluorophenylamino)-4-(2-hydroxy-1,2,2-triphenylethylamino)cyclobut-3-ene-1,2-dione (5bb)
Following the general procedure, compound 5bb was obtained after 62 h of reaction at room temperature as a white solid in >95% yield. [α]27D = −197 (c 0.65, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 10.12 (br s, 1H), 8.73 (br d, J = 9.7 Hz, 1H), 7.6 (d, J = 7.5 Hz, 2H), 7.35 (t, J = 7.6 Hz, 2H), 7.25–7.05 (m, 13H), 6.85–6.79 (m, 1H), 6.69 (br s, 1H), 6.23 (br d, J = 9.8 Hz, 1H). 13C APT-NMR (100 MHz, DMSO-d6) δ 184.5 (1C), 179.7 (1C), 168.1 (1C), 162.9 (d, J = 242.5 Hz, 1C), 162.8 (1C), 162.7 (d, J = 232.5 Hz, 1C), 145.6 (1C), 144.1 (1C), 141.5 (t, J = 13.7 Hz, 1C), 139.0 (1C), 128.9 (2C), 128.0 (2C), 127.4 (4C) 127.0 (1C), 126.7 (1C), 126.5 (1C), 126.2 (2C), 126.1 (2C), 101.2 (d, J = 29.5 Hz, 2C), 97.4 (t, J = 26.5 Hz, 1C) 80.4 (1C), 63.3 (1C). IR (KBr film) (cm−1) ν 3483, 3446, 3227, 3122, 2924, 2853, 1800, 1656, 1608, 1565, 1493, 1465, 1377, 1188, 1166, 1119, 1065, 1004, 891, 761, 750, 728, 697. MS (ESI) 497.2 [M + H].(R)-3-(3,5-Bis(trifluoromethyl)phenylamino)-4-(1-phenylethylamino)cyclobut-3-ene-1,2-dione (5ac)
Following the general procedure, compound 5ac was obtained after 85 h of reaction at room temperature as a white solid in 84% yield. [α]27D = −6 (c 0.87, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 10.06 (br s, 1H), 8.15 (br s, 1H), 8.01 (s, 2H), 7.67 (s, 1H), 7.44–7.30 (m, 5H), 5.36–5.27 (m, 1H), 1.61 (d, J = 6.9 Hz, 3H). 13C APT-NMR (100 MHz, DMSO-d6) δ 184.3 (1C), 180.5 (1C), 168.8 (1C), 162.5 (1C), 142.7 (1C), 140.9 (1C), 131.2 (q, J = 34.0 Hz, 2C), 128.6 (2C), 127.5 (1C), 126.0 (2C), 123.0 (q, J = 271.2 Hz, 2C), 118.0 (2C), 114.7 (1C), 53.3 (1C), 22.7 (1C). IR (KBr film) (cm−1) ν 3135, 2924, 1797, 1661, 1570, 1559, 1491, 1456, 1377, 1275, 1188, 1170, 1129, 944, 883, 750, 697, 683, 667. MS (ESI+) 429.1 [M + H].(R)-3-(3,5-Bis(trifluoromethyl)phenylamino)-4-(1-hydroxy-3-phenylpropan-2-ylamino)cyclobut-3-ene-1,2-dione (5ad)
Following the general procedure, compound 5ad was obtained after 96 h of reaction at room temperature as a white solid in 60% yield. [α]24D = +125 (c 0.54, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 9.78 (br s, 1H), 7.59 (s, 2H), 7.47 (br d, J = 9.0 Hz, 1H), 7.19 (s, 1H), 6.85–6.73 (m, 5H), 4.78 (br s, 1H), 3.90–3.80 (m, 1H), 3.15–3.06 (m, 2H), 2.51 (dd, J = 6.6, 13.6 Hz, 1H), 2.38 (dd, J = 7.9, 13.4 Hz, 1H). 13C APT-NMR (100 MHz, DMSO-d6) δ 184.2 (1C), 180.1 (1C), 169.5 (1C), 162.0 (1C), 141.1 (1C), 137.8 (1C), 131.3 (q, J = 33.6 Hz, 2C), 129.2 (2C), 128.2 (2C), 126.2 (1C), 123.1 (q, J = 270.9 Hz, 2C), 117.7 (2C), 114.5 (1C), 62.2 (1C), 57.4 (1C), 38.0 (1C). IR (KBr film) (cm−1) ν 3280, 3044, 2923, 2853, 1790, 1690, 1605, 1576, 1553, 1499, 1477, 1455, 1388, 1280, 1181, 1171, 1131, 1030, 899, 882, 701. HRMS (ESI+) calcd C21H17F6N2O3 459.1144; found 459.1130 [M + H].3-(3,5-Bis(trifluoromethyl)phenylamino)-4-((1S,2R)-2-hydroxy-2,3-dihydro-1H-inden-1-ylamino)cyclobut-3-ene-1,2-dione (5ae)
Following the general procedure, compound 5ae was obtained after 52 h of reaction at room temperature as a white solid in 61% yield. [α]27D = +81 (c 0.48, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 10.46 (br s, 1H), 8.16 (br d, J = 9.0 Hz, 1H), 8.10 (s, 2H), 7.65 (s, 1H), 7.32–7.22 (m, 4H), 5.62 (br s, 1H), 5.53 (dd, J = 5.0, 8.7 Hz, 1H), 4.65–4.55 (m, 1H), 3.16 (dd, J = 4.8, 16.4 Hz, 1H), 2.89 (d, J = 16.2 Hz, 1H). 13C APT-NMR (100 MHz, DMSO-d6) δ 184.6 (1C), 180.4 (1C), 169.5 (1C), 162.6 (1C), 141.2 (1C), 141.2 (1C), 140.5 (1C), 131.3 (q, J = 32.7 Hz, 2C), 128.0 (2C), 126.6 (1C), 125.1 (1C), 124.2 (1C), 123.1 (q, J = 271.1 Hz, 2C), 117.8 (2C), 114.5 (1C), 72.2 (1C), 62.2 (1C). IR (KBr film) (cm−1) ν 3278, 2958, 2927, 2854, 1792, 1691, 1597, 1552, 1482, 1465, 1442, 1383, 1334, 1300, 1190, 1128, 1043, 933, 897, 742, 685. MS (ESI+) 457.1 [M + H].3-(3,5-Difluorophenylamino)-4-((1S,2R)-2-hydroxy-2,3-dihydro-1H-inden-1-ylamino)cyclobut-3-ene-1,2-dione (5be)
Following the general procedure, compound 5be was obtained after 40 h of reaction at room temperature as a white solid in 88% yield. [α]27D = +48 (c 0.47, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 10.16 (br s, 1H), 8.17 (br d, J = 9.4 Hz, 1H), 7.32–7.23 (m, 6H), 6.85 (t, J = 9.5 Hz, 1H), 5.58 (br s, 1H), 5.52 (dd, J = 5.0, 9.0 Hz, 1H), 4.60–4.56 (m, 1H), 3.14 (dd, J = 4.0, 15.7 Hz, 1H), 2.87 (d, J = 16.3 Hz, 1H). 13C APT-NMR (100 MHz, DMSO-d6) δ 184.4 (1C), 180.2 (1C), 169.3 (1C), 163.0 (d, J = 238.2 Hz, 1C), 162.8 (1C), 162.9 (d, J = 242.5 Hz, 1C), 141.9 (t, J = 13.7 Hz, 1C), 141.1 (1C), 140.5 (1C), 128.0 (1C), 126.6 (1C), 125.1 (1C), 124.2 (1C), 101.0 (d, J = 29.4 Hz, 2C), 97.2 (d, J = 26.1 Hz, 1C), 72.3 (1C), 61.2 (1C), 39.4 (1C). IR (KBr film) (cm−1) ν 3460, 3261, 2922, 2852, 1794, 1662, 1613, 1576, 1559, 1541, 1446, 1416, 1377, 1208, 1149, 1119, 1090, 1022, 995, 977, 854, 752, 727, 686, 646. MS (ESI+) 357.1 [M + H].(S)-3-(3,5-Difluorophenylamino)-4-(1-(naphthalen-1-yl)ethylamino)cyclobut-3-ene-1,2-dione (5bf)
Following the general procedure, compound 5bf was obtained after 33 h of reaction at room temperature as a white solid in 82% yield. [α]27D = +184 (c 0.44, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 9.80 (br s, 1H), 8.28 (br d, J = 6.9 Hz, 1H), 8.14 (d, J = 8.4 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.67–7.55 (m, 4H), 7.16 (br d, J = 7.6 Hz, 2H), 6.84 (t, J = 9.3 Hz, 1H), 6.17–6.08 (m, 1H), 1.75 (d, J = 6.7 Hz, 3H). 13C APT-NMR (100 MHz, DMSO-d6) δ 184.1 (1C), 180.1 (1C), 168.4 (1C), 162.9 (d, J = 242.5 Hz, 1C), 162.8 (1C), 162.8 (d, J = 242.5 Hz, 1C), 141.6 (t, J = 13.7 Hz, 1C), 138.1 (1C), 133.4 (1C), 129.9 (1C), 128.7 (1C), 128.2 (1C), 126.7 (1C), 125.9 (1C), 125.4 (1C), 122.7 (1C), 122.6 (1C), 101.1 (d, J = 29.4 Hz, 2C), 97.4 (t, J = 26.2 Hz, 1C), 49.4 (1C), 22.7 (1C). IR (KBr film) (cm−1) ν 3165, 3080, 3017, 2924, 2854, 1793, 1664, 1631, 1609, 1460, 1375, 1307, 1153, 1116, 993, 854, 835, 804, 782, 754. MS (ESI+) 379.2 [M + H].3-((1R,2R)-2-(Piperidin-1-yl)cyclohexylamino)-4-(3,4,5-trifluorophenylamino)cyclobut-3-ene-1,2-dione (5fg)
Following the general procedure, compound 5fg was obtained after 69 h of reaction at room temperature as a white solid in 82% yield. [α]25D = −54 (c 0.40, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 9.92 (br s, 1H), 7.52 (br s, 1H), 7.33 (d, J = 9.7 Hz, 1H), 7.31 (d, J = 9.5 Hz, 1H), 4.02–3.85 (m, 1H), 2.76–2.56 (m, 2H), 2.39–2.17 (m, 3H), 2.10–1.97 (m, 1H), 1.91–1.80 (m, 1H), 1.79–1.60 (m, 2H), 1.51–1.00 (m, 10H). 13C APT-NMR (100 MHz, DMSO-d6) δ 184.5 (1C), 180.1 (1C), 169.6 (1C), 161.7 (2C), 150.6 (dd, J = 10.0, 245.8 Hz, 1C), 150.5 (dd, J = 10.0, 244.7 Hz, 1C), 134.3 (dt, J = 15.6, 243.0 Hz, 1C), 102.5 (d, J = 23.7 Hz, 2C), 68.3 (1C), 54.3 (1C), 49.2 (1C), 33.6 (1C), 26.2 (1C), 24.6 (2C), 24.4 (1C), 24.3 (1C), 23.2 (2C). IR (KBr film) (cm−1) ν 3194, 3084, 2924, 2853, 1796, 1657, 1631, 1575, 1480, 1450, 1377, 1241, 1154, 1098, 1044, 980, 856, 799, 744, 462. MS (ESI+) 408.2 [M + H].3-((R)-2′-Hydroxy-1,1′-binaphthyl-2-ylamino)-4-((1R,2R)-2-(piperidin-1-yl)cyclohexylamino)cyclobut-3-ene-1,2-dione (5hg)
Following the general procedure, compound 5hg was obtained after 88 h of reaction at room temperature as a yellow solid in 68% yield. [α]24D = +85 (c 0.44, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.01 (d, J = 8.8 Hz, 1H), 7.92–7.79 (m, 3H), 7.62 (br d, J = 6.7 Hz, 1H), 7.40 (dt, J = 1.1, 8.0 Hz, 1H), 7.34–7.11 (m, 5H), 6.96 (d, J = 8.3 Hz, 1H), 6.00 (br s, 2H), 3.85 (br s, 1H), 2.77–2.10 (m, 6H), 1.91–1.51 (m, 3H), 1.49–0.99 (m, 11H). 13C APT-NMR (75 MHz, CDCl3) δ 184.1 (1C), 182.1 (1C), 168.4 (1C), 163.8 (1C), 152.7 (1C), 134.5 (1C), 133.5 (1C), 133.4 (1C), 131.53 (1C), 130.7 (1C), 130.1 (1C), 129.0 (2C), 128.2 (2C), 127.4 (1C), 127.1 (1C), 125.7 (1C), 125.5 (1C), 124.1 (1C), 123.5 (1C), 120.7 (1C), 118.8 (1C), 113.2 (1C), 67.9 (1C), 54.2 (1C), 49.5 (2C), 34.8 (1C), 25.2 (1C), 24.9 (1C), 24.2 (2C), 23.8 (1C), 23.2 (1C). IR (KBr film) (cm−1) ν 3251, 2924, 2854, 1792, 1683, 1595, 1505, 1463, 1377, 1338, 1261, 1095, 1021, 801. MS (ESI+) 546.3 [M + H].3-((R)-2′-Hydroxy-1,1′-binaphthyl-2-ylamino)-4-((1R,2R)-2-(pyrrolidin-1-yl)cyclohexylamino)cyclobut-3-ene-1,2-dione (5hh)
Following the general procedure, compound 5hh was obtained after 92 h of reaction at room temperature as a yellow solid in 50% yield. [α]24D = +227 (c 0.48, CHCl3). 1H NMR (400 MHz, CDCl3) 7.96 (d, J = 8.8 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.81 (app. dd, J = 6.7, 7.5 Hz, 2H), 7.70–7.53 (m, 1H), 7.40 (t, J = 7.4 Hz, 1H), 7.33–7.09 (m, 5H), 6.94 (d, J = 8.4 Hz, 1H), 6.32 (br s, 2H), 3.72 (br s, 1H), 2.75–2.45 (m, 4H), 2.12–1.87 (m, 1H), 1.80–1.40 (m, 4H), 1.39–0.73 (m, 9H). 13C APT-NMR (100 MHz, CDCl3) δ 183.7 (1C), 181.9 (1C), 168.5 (1C), 164.8 (1C), 153.1 (1C), 134.5 (1C), 133.6 (1C), 133.3 (1C), 131.5 (1C), 130.4 (1C), 129.5 (1C), 128.8 (2C), 128.2 (1C), 128.1 (1C), 127.1 (1C), 126.9 (1C), 125.9 (1C), 125.5 (1C), 124.2 (1C), 123.3 (1C), 121.7 (1C), 119.2 (1C), 114.1 (1C), 63.1 (1C), 55.8 (1C), 48.0 (2C), 34.3 (1C), 29.7 (1C), 24.2 (1C), 24.0 (1C), 23.5 (2C). IR (KBr film) (cm−1) ν 3240, 2924, 2854, 1792, 1683, 1596, 1505, 1462, 1428, 1377, 1336, 1271, 815, 748. MS (ESI+) 532.3 [M + H].Acknowledgements
We thank the Lilly Company for the concession of the Spanish Lilly Prize 2012 to R. P. Herrera, which has allowed this research. J.V.A.-R. thanks the DGA for a predoctoral fellowship. We also thank Prof. M. Concepción Gimeno (ISQCH) for her help and encouragement in our research.Notes and references
- (a) G. H. Posner, Chem. Rev., 1986, 86, 831 CrossRef CAS; (b) P. A. Dalby, G. J. Lye and J. M. Woodley, in Handbook of Chiral Chemicals, ed. D. Ager, CRC, Boca Raton, 2005, pp. 419–428 Search PubMed; (c) C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605 CrossRef CAS PubMed; (d) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef PubMedand references therein cited. For some interesting examples, see also: (e) A. Saha, R. B. N. Baig, J. Leazer and R. S. Varma, Chem. Commun., 2012, 48, 8889 RSC; (f) R. B. N. Baig and R. S. Varma, Green Chem., 2012, 14, 625 RSC; (g) R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2014, 16, 2122 RSC.
- A. M. Walji and D. W. C. MacMillan, Synlett, 2007, 1477 CAS.
- For pivotal reviews in this area of research, see: (a) J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem.–Eur. J., 2011, 17, 6890 CrossRef PubMed; (b) R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC; (c) F. R. Wurm and H.-A. Klok, Chem. Soc. Rev., 2013, 42, 8220 RSC; (d) J. V. Alegre-Requena, Synlett, 2014, 298 CASFor selected examples, see: (e) P. Sejwal, Y. Han, A. Shah and Y.-Y. Luk, Org. Lett., 2007, 9, 4897 CrossRef CAS PubMed; (f) Ł. Albrecht, G. Dickmeiss, C. F. Weise, C. Rodríguez-Escrich and K. A. Jørgensen, Angew. Chem., Int. Ed, 2012, 51, 13109 CrossRef PubMed; (g) K. S. Yang, A. E. Nibbs, Y. E. Türkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 16050 CrossRef CAS PubMed; (h) J. V. Alegre-Requena, E. Marqués-López, P. J. Sanz Miguel and R. P. Herrera, Org. Biomol. Chem., 2014, 12, 1258 RSC.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS PubMed.
- (a) B. M. Trost, Science, 1991, 254, 1471 CAS; (b) B. M. Trost, Acc. Chem. Res., 2002, 35, 695 CrossRef CAS PubMed.
- (a) E. Marqués-López, J. V. Alegre-Requena and R. P. Herrera, European Pat. EP14382260.9 filed, July 07, 2014; (b) F. Wurm, T. Steinbach and H.-A. Klok, Chem. Commun., 2013, 49, 7815 RSC ; Although the authors in this article argue a possible one-pot strategy for bioconjugation, in the experimental procedure detailed in the supplementary information of this work, the authors take a portion of a first reaction (in phosphate buffer) from a reactor and they pour it into another reactor where the second synthetic step takes place (in borate buffer), which is not a strict one-pot reaction.
- C. Reichardt and T. Welton, in Solvents and Solvent Effects in Organic Chemistry, ed. C. Reichardt, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 4th edn, 2003 Search PubMed.
- S. Cohen and S. G. Cohen, J. Am. Chem. Soc., 1966, 88, 1533 CrossRef CAS.
- H. Konishi, T. Y. Lam, J. P. Malerich and V. H. Rawal, Org. Lett., 2010, 12, 2028 CrossRef CAS PubMed.
- For the preparation of the opposite enantiomer, see: W. Yang and D.-M. Du, Adv. Synth. Catal., 2011, 353, 1241 CrossRef CAS.
- Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CrossRef CAS PubMed.
- W. Yang and D.-M. Du, Org. Lett., 2010, 12, 5450 CrossRef CAS PubMed.
- H. Jiang, M. W. Paixão, D. Monge and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 2775 CrossRef CAS PubMed.
- For the second step (40% yield), see: R. Baran, E. Veverková, A. Škvorcová and R. Šebesta, Org. Biomol. Chem., 2013, 11, 7705 CAS The authors did not detail the yield of the first step, we thus took the yield of the first step showed in Rawal's work (85% yield) (ref. 4).
- For the preparation of the opposite enantiomer, see: W. Yang and D.-M. Du, Org. Biomol. Chem., 2012, 10, 6876 CAS.
- F. Olmo, C. Rotger, I. Ramírez-Macías, L. Martínez, C. Marín, L. Carreras, K. Urbanová, M. Vega, G. Chaves-Lemaur, A. Sampedro, M. J. Rosales, M. Sánchez-Moreno and A. Costa, J. Med. Chem., 2014, 57, 987 CrossRef CAS PubMed.
- (a) P. M. M. Guedes, G. K. Silva, F. R. S. Gutierrez and J. S. Silva, Expert Rev. Anti-Infect. Ther., 2011, 9, 609 CrossRef PubMed; (b) J. A. Urbina, Acta Trop., 2010, 115, 55 CrossRef PubMed; (c) H. Cerecetto and M. González, Pharmaceuticals, 2010, 3, 810 CrossRef CAS PubMed.
- M. J. Coghlan, W. A. Carroll and M. Gopalakrishnan, J. Med. Chem., 2001, 44, 1627 CrossRef CAS.
- F. M. Ashcroft and F. M. Gribble, Trends Neurosci., 1998, 21, 288 CrossRef CAS.
- For the pioneering syntheses, see: (a) J. A. Butera and S. A. Antane, US Pat. 5,506,252 filed, Oct. 2, 1994, and issued April 9, 1996; (b) J. A. Butera, M. M. Antane, S. A. Antane, T. M. Argentieri, C. Freeden, R. F. Graceffa, B. H. Hirth, D. Jenkins, J. R. Lennox, E. Matelan, N. W. Norton, D. Quagliato, J. H. Sheldon, W. Spinelli, D. Warga, A. Wojdan and M. Woods, J. Med. Chem., 2000, 43, 1187 CrossRef CAS PubMed.
- For selected studies using WAY-133537 (14), see: (a) A. Wojdan, C. Freeden, M. Woods, G. Oshiro, W. Spinelli, T. J. Colatsky, J. H. Sheldon, N. W. Norton, D. Wargan, M. M. Antane, S. A. Antane, J. A. Butera and T. M. Argentieri, J. Pharmacol. Exp. Ther., 1999, 289, 1410 CAS; (b) S. A. Buckner, I. Milicic, A. V. Daza, M. J. Coghlan and M. Gopalakrishnan, Br. J. Pharmacol., 2002, 135, 639 CrossRef CAS PubMed; (c) A. C. Fabiyi, M. Gopalakrishnan, J. J. Lynch III, J. D. Brioni, M. J. Coghlan and M. E. Brune, BJU Int., 2003, 91, 284 CrossRef CAS; (d) J. A. Butera, D. J. Jenkins, J. R. Lennox, J. H. Sheldon, N. W. Norton, D. Warga and T. M. Argentieri, Bioorg. Med. Chem. Lett., 2005, 15, 2495 CrossRef CAS PubMed; (e) R. J. Altenbach, M. E. Brune, S. A. Buckner, M. J. Coghlan, A. V. Daza, A. Fabiyi, M. Gopalakrishnan, R. F. Henry, A. Khilevich, M. E. Kort, I. Milicic, V. E. Scott, J. C. Smith, K. L. Whiteaker and W. A. Carroll, J. Med. Chem., 2006, 49, 6869 CrossRef CAS PubMed.
- For selected studies using WAY-151616 (18), see: (a) M. M. Antane, D. R. Herbst, G. R. McFarlane, E. G. Gundersen, B. H. Hirth, D. A. Quagliato, R. F. Graceffa, J. A. Butera and A. M. Gilbert, Interanational Pat. WO 98/02413 filed Jul. 16, 1997 and issued, June 22, 1998; (b) M. J. Gast and T. R. Koziol, Interanational Pat. WO 98/11888 filed Sept. 17, 1997 and issued, March 26, 1998; (c) J. A. Butera and T. M. Argentieri, Drugs Future, 2000, 25, 239 CrossRef CAS; (d) J.-N. Mahy Gehenne, M. J. Rodriguez Allue and M. Pugliese, Interanational Pat. WO 2006/000607 filed Jun. 23, 2005 and issued, January 5, 2006.
- (a) D. R. Herbst, M. M. Antane, G. R. McFarlane, E. G. Gundersen, B. H. Hirth, D. A. Quagliato, R. F. Graceffa and J. A. Butera, US Pat. 5,763,474 filed Jul. 7, 1997 and issued, June 9, 1998; (b) A. M. Gilbert, M. M. Antane, T. M. Argentieri, J. A. Butera, G. D. Francisco, C. Freeden, E. G. Gundersen, R. F. Graceffa, D. Herbst, B. H. Hirth, J. R. Lennox, G. McFarlane, N. W. Norton, D. Quagliato, J. H. Sheldon, D. Warga, A. Wojdan and M. Woods, J. Med. Chem., 2000, 43, 1203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental information, 1H and 13C NMR spectra for all new squaramides 5. See DOI: 10.1039/c5ra05383h |
| This journal is © The Royal Society of Chemistry 2015 |