
Targeted killing of metastatic cells using a platelet-inspired drug delivery system
Victor Pan,
Preethi N. Siva,
Christa L. Modery-Pawlowski,
Ujjal Didar Singh Sekhon and
Anirban Sen Gupta*
Department of Biomedical Engineering, Case Western Reserve University, 2071 Martin Luther King Drive, Wickenden 517B, Cleveland, Ohio 44106, USA. E-mail: axs262@case.edu; Fax: +1 216-368-4969; Tel: +1 216-368-4564
First published on 12th May 2015
Abstract
The ‘targeted nanomedicine’ approach has revolutionized cancer therapy by packaging drugs within nanovehicles that can accumulate preferentially within the tumor due to the ‘enhanced permeation and retention’ (EPR) mechanism (passive targeting) and further bind to cancer cells via specific ligand–receptor interactions (active targeting). While these approaches have shown promise in well-vascularized primary tumors, their use for metastatic sites faces challenges due to high heterogeneity in the expression levels of tumor-associated targetable receptors. Therefore, alternative strategies for metastatic cell-specific targeting are of great clinical interest. To this end, we are exploring binding interactions of metastatic cells with host cells in the vascular compartment and whether such interactions can be exploited for metastasis-targeted delivery of drug-loaded nanovehicles. For ‘proof-of-concept’ of this approach, using high-metastatic MDA-MB-231 versus low-metastatic MCF-7 human breast cancer cells, we demonstrate that high-metastatic cells have enhanced binding interactions with active platelets via several cell-surface proteins. Utilizing this information and using liposomes as a model nanovehicle, we have created delivery systems that can actively bind to the high-metastatic cells via platelet-inspired heteromultivalent interactions. Using a mono-culture of MCF-7 versus MDA-MB-231, we demonstrate that these systems enhance the delivery of Doxorubicin (DOX) to the high-metastatic cells to cause significant cell-killing. Furthermore, incubating co-cultures of MCF-7 and MDA-MB-231 cells with the DOX-loaded platelet-inspired nanovehicles, we demonstrate that the targeted binding, DOX delivery and cell-killing are quite selective towards the high-metastatic cells. Our results suggest that this platelet-inspired targeting can lead to unique strategies for metastasis-selective nanomedicine.
1. Introduction
Cancer metastasis, i.e. spreading of cancer from a primary site to distal tissues, is the major cause of cancer-related mortalities.1 In recent years, significant technological advances in early cancer detection and focused surgical and therapeutic strategies have substantially improved the survival of patients with primary tumors. However, survival statistics of patients with metastatic cancer remains poor due to challenges of efficient detection and treatment of cancer that has spread to multiple sites. The current clinical mainstay for treating metastasis is systemic administration of therapeutic agents (chemotherapy, biologics, hormone therapy, etc.), with the expectation that such systemic delivery will lead to some fraction of the dose reaching and acting on the metastatic sites.2,3 During the past decade, advances in the ‘nanomedicine’ approach has provided an improvement in cancer treatment, by packaging therapeutic agents within injectable nanoparticles that are too big to pass through the endothelial junctions of healthy tissue vasculature or to be cleared rapidly by the kidney, but small enough to permeate through the fenestrated leaky vasculature associated with tumors.4–8 Once localized within the tumor stroma, these drug-loaded nanoparticles stay retained for sustained periods of time due to compromised lymphatic drainage of the tumor and thus act as accumulated drug delivery depots. This mechanism is now famously known as the Enhanced Permeation and Retention (EPR) effect for ‘passive targeting’ of nanoparticles (and drug–macromolecule conjugates).9 This mechanisms is utilized in several clinical chemotherapeutic formulations like Doxil®, Duanosome®, Myocet® and Lipoplatin® to enhance preferential uptake of the drugs in the tumor tissue while reducing systemic non-specific uptake and toxicity.5 Beyond EPR-based ‘passive targeting’ mechanism, the ‘nanomedicine’ approach has further progressed into utilizing ‘active targeting’, where the drug-loaded nanovehicles are surface-decorated with ligands that facilitate active binding to receptors uniquely expressed or spatio-temporally upregulated on cancer cells, to provide a molecular mechanism of cell-specific nanovehicle anchorage and internalization (e.g. via receptor-mediated endocytosis).10–12 A partial example of this approach in clinically approved nanomedicine is found in Abraxane®, an albumin nanoparticle formulation of paclitaxel that is thought to utilize the specific binding of albumin to GP60 receptors on tumor vasculature-associated endothelial cells and undergo transcytosis into the tumor stroma.13A large variety of ‘actively targeted nanomedicine’ formulations are currently under extensive pre-clinical evaluation for different cancers. However, the majority of these studies involve animal models of primary tumors, and in the few selective studies that involve metastatic cancer models, the nanomedicine targeting mechanism to metastasis is merely an extension of the same EPR and active targeting mechanisms that are being tried for the primary tumor. This approach can be problematic in metastasis-specific targeting because (i) the metastatic sites may not have a sufficiently vascularized environment to allow sufficient EPR mechanism14–17 and (ii) the spatio-temporal expression of tumor-associated cell-surface receptors conventionally used for active targeting (e.g. Epidermal Growth Factor Receptors, Folate Receptors, Transferrin Receptors, etc.) is highly heterogeneous in metastatic tumors compared to primary tumors.18–21 Therefore, there is a significant interest in alternative strategies for specific targeting of nanovehicles to metastatic cells. To this end, we are investigating the molecular interactions of metastatic cells with host cells, especially in the vascular compartment during hematologic pathways of metastasis. An interesting component in this context is the interaction of metastatic cells with platelets during dissemination from the primary site, intravasation, circulation and adhesion/extravasation at the distal site.
Disseminated metastatic cells that intravasate into circulation (sometimes referred to as circulating tumor cells or CTCs) are expected to have poor survival rates due to lack of matrix adhesion, increased fluid mechanical stresses of blood circulation and action of immune cells.22–25 Yet, some of these cells are found to effectively avoid anoikis, undergo epithelial-to-mesenchymal transformation en route to intravasation, avoid immune surveillance in circulation and undergo arrest or adhesion at distal site vasculature to form metastatic colonies.26,27 Several research studies in the past and in recent years have demonstrated that active platelets and platelet-derived secretome may play a significant mechanistic role in facilitating components these processes, for example, by promoting epithelial-to-mesenchymal transformation in cancer cells by secretion of TGFβ and activation of TGFβ/Smad and NF-kB pathways, secreting pro-angiogenic cytokines like VEGF-A and PDGF, secreting matrix degrading proteases to help in intravasation of CTCs into blood circulation, forming a ‘microthrombotic cloak’ around CTCs to help avoid surveillance and action by immune cells in circulation, and mediating adhesion onto the vascular wall at distal sites followed by extravasation and pro-metastatic activities.28–35 Fig. 1A shows a schematic of platelet-facilitated dissemination/intravasation, hematologic transport, immune-avoidance and distal site adhesion/extravasation of CTCs, while Fig. 1B shows possible binding interactions of CTCs with active platelets and vascular endothelium. Interestingly, there have been clinical studies regarding investigation of anti-platelet and anti-coagulatory agents for therapeutic benefit in metastatic patients.36 However, these studies have shown only limited success, possibly due to other systemic side-effects of such drugs. Rationalizing from these research findings, we have hypothesized that a strategy that does not systemically alter platelet function but rather exploits the molecular interactions between active platelets and metastatic cells for cell-specific binding, can provide a unique alternative way for active targeting of nanovehicles to metastatic cells.
 | ||
| Fig. 1 [A] Schematic representation of platelet involvement in facilitating mechanisms of (1) dissemination/intravasation of CTCs, (2) binding to CTCs in hematologic transport, (3) immune-avoidance of CTCs via microthrombi ‘cloak’ formation and (4) distal site adhesion/extravasation of CTCs leading to pro-metastatic activities; [B] shows possible ligand-receptor mediated binding interactions of CTCs with active platelets and vascular endothelium. | ||
To this end, as ‘proof-of-concept’ we have compared the interaction of human platelets with the low-metastatic human breast cancer cell line MCF-7 versus the high-metastatic human breast cancer cell line MDA-MB-231 in a flow environment in vitro, have identified receptor expressions on these cell lines that can possibly mediate platelet-interactivity, and have utilized this information to develop heteromultivalently surface-engineered nanoconstructs using liposomes as a model vehicle. These nanovehicles can exploit platelet-inspired interactivity to bind selectively to high-metastatic cells. We previously demonstrated that such nanovehicles labeled with a model fluorescent molecule (Rhodamine-B) were able to bind the MDA-MB-231 cells significantly more compared to MCF-7 cells.37 Building on this work, here we explored the ability of these nanovehicles to deliver a clinically relevant chemotherapeutic drug Doxorubicin (DOX) to high-metastatic MDA-MB-231 cells compared to low-metastatic MCF-7 cells. First using mono-cultures and subsequently using co-cultures of the low-metastatic and the high-metastatic cells, we demonstrate that the platelet-inspired nanovehicles render enhanced binding and resultant delivery DOX preferentially more to the high-metastatic cells in vitro, resulting in increased killing of these cells compared to the low-metastatic ones.
2. Materials and methods
2.1. Materials
MDA-MB-231 and MCF-7 cells were purchased from American Type Culture Collection (Manassas, VA). RPMI 1640 cell culture media, penicillin, streptomycin, Dulbecco's phosphate buffered saline (DPBS), phosphate buffered saline (PBS), sodium carbonate, 0.25% Trypsin, paraformaldehyde (PFA) were purchased from Fisher Scientific (Pittsburgh, PA). Calcein AM AlexaFluor 488, CellTrace™ Carboxyfluorescein Succinimidyl Ester (CFSE), CellTrace™ Violet and 4′,6-diamidino-2-phenylidole (DAPI) were purchased from Life Technologies (Grand Island, NY). Adenosine Diphosphate (ADP) was purchased from Bio/Data Corporation (Horsham, PA). Distearyl phosphatidyl choline (DSPC), carboxy-polyethylene glycol-modified distearyl phosphatidyl ethanolamine (DSPE-PEG2000-COOH), were purchased from Avanti Polar Lipids (Miami, FL). Cholesterol, Triton-X100, sodium carbonate, and sodium chloride were purchased from Sigma-Aldrich (St. Lous, MO). Resins and amino acids for peptide synthesis were purchased from Advanced ChemTech (Louisville, KY), and Doxorubicin–HCl from LC Laboratories (Woburn, MA). Fluorescein isothiocyanate (FITC)-conjugated anti-CD41a was purchased from BioLegend (San Diego, CA). Phycoerythin (PE)-conjugated anti-CD62P, CD62E, CD62L, and CD42b and FITC-conjugated anti-CD51/61 and CD49b were purchased from BD Bioscience (San Jose, CA). All targeting peptides were synthesized on Knorr resin using solid phase peptide chemistry technique, and subsequently conjugated via their amino termini to the carboxyl termini of DSPE-PEG2000-COOH.382.2. Cell culture
Human breast cancer cell lines, MCF-7 (low metastatic) and MDA-MB-231 (high metastatic) were grown in RPMI 1640 medium supplemented with 10% FBS, penicillin (50 units per mL), and streptomycin (50 μg mL−1). All cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C for all experiments.2.3. Interaction of cancer cells with human platelets and subsequent analysis of relevant antigen expression on the cancer cells that mediate such interactions
Since metastatic cells like MDA-MB-231 are known to spread through blood circulation and adhere to vascular walls at distal sites,38 we looked at the innate ability of these high-metastatic (MDA-MB-231) cancer cells versus low-metastatic (MCF-7) cancer cells to interact with human platelets. For this, MDA-MB-231 and MCF-7 cells were cultured in black-walled well plates until ∼80% confluent, media was removed, and the cells were gently washed with 1× DPBS. Cells were subsequently fixed with 4% PFA for 15 min at 37 °C, and then washed with 1× DPBS to remove excess fixative. The cells were then incubated with 0.1% Triton-X100 in 1× DPBS for 1 min to transiently permeabilize the cell membrane, and Phalloidin at 6 μM (λex = 650 nm, λem = 668 nm) in 1× DPBS was added to stain the F-actin cytoskeleton of the cancer cells for 15 min at room temperature. Following this, the nuclei of the cells were stained with DAPI at 300 nM in 1× PBS (λex = 358 nm, λem = 461 nm) for 5 min at room temperature. Platelet-rich plasma (PRP) was obtained from human whole blood by centrifugation (150 g, 15 min, room temperature). 10 μl of calcein (λex = 495 nm, λem = 515 nm) in 1× DPBS was added for every 5 mL of PRP, and allowed to incubate for 20 minutes at 37 °C to stain the platelet cytoplasm with green fluorescence. Next, the Calcein stained PRP was centrifuged at 2500 g for 25 min at room temperature to produce a platelet pellet and supernatant platelet-poor plasma (PPP) was removed to get rid of excess Calcein. The platelet pellet was resuspended in fresh PPP and incubated with ADP at a concentration of 2 × 10−5 M for 30 min at 37 °C. The Calcein-stained activated platelets in PPP were then allowed to incubate with the cancer cells in the well plates for 30 min at 37 °C, in the shaker incubator at 100 rpm, which produces a surface shear of ∼3 dynes cm−2 (low arterial and large vein shear range).39 Following this, any unbound (and loosely bound) platelets were washed off with 1× DPBS, and platelets stably bound to the cancer cells were imaged using a Zeiss inverted fluorescence microscope utilizing appropriate filters for Phalloidin, DAPI and Calcein fluorescence, to obtain fluorescence co-localization images.Observation of the results from the above experiments led to analysis of cell-surface receptors and antigens on the MDA-MB-231 and MCF-7 cells, especially in relevance to collagen-interactions and platelet-binding. For this, the cancer cells were cultured in black-walled well plates as before, fixed with 4% PFA for 15 min at room temperature, and then incubated with 10% BSA in 1× DPBS for 1 h at room temperature to block any non-specific antibody binding. Cells were then incubated with several different fluorescently (FITC, λex = 490 nm, λem = 525 nm) labeled antibodies as shown in Fig. 2C for 1 h at room temperature in the dark. This group of antibodies was selected based upon the knowledge of antigens that are present on the natural platelet surface that help platelet interactions with cells and vascular matrix proteins. We rationalized that this group was an appropriate initial metric in order to analyze platelet-mimetic and platelet-relevant interactions on the cancer cells. Following incubation, unbound antibody was removed and rinsed with 1× DPBS, and antibody associated fluorescence was measured using a fluorescence plate reader to obtain fluorescence intensity levels. These levels were compared between MCF-7 and MDA-MB-231 cells, and the results were interpreted as the extent of receptor expression (‘++ = abundant’, ‘+ = slightly abundant’, ‘− = non-abundant’ in Fig. 2B).
 | ||
| Fig. 2 Top panel shows representative multispectral fluorescence images of calcein-stained (green) resting versus active (ADP-activated) platelets binding to phalloidin (red cytoskeleton) and DAPI (blue nucleus) co-stained MCF-7 or MDA-MB-231 cells; the resting platelets show minimal binding to both cell lines, while the active platelets show a significantly enhanced binding to MDA-MB-231 cells compared to MCF-7 (all images are at same magnification and the scale bar shown on the MCF-7 image represents 20 μm). Please note the morphological difference between the low metastatic MCF-7 (roundish morphology) versus high metastatic MDA-MB-231 (elongated morphology). Bottom panel table shows immunofluorescence-assisted estimation of relative expression levels of possible platelet-relevant and platelet-interactive receptors on MCF-7 cells versus MDA-MB-231 cells, with ‘++’ representing ‘abundant’, ‘+’ representing ‘slightly abundant’, and ‘−’ representing ‘non-abundant’. | ||
Based on the receptor antigen analysis data, additional experiments were carried out by modifying the surface of glass slides with appropriate peptide ligands that can bind to the ‘abundant’ receptor antigens of the cancer cells and allowing DAPI-stained (blue nucleus) MCF-7 or MDA-MB-231 cells to flow over these modified glass slide surfaces at a wall shear stress of 5 dyn cm−2. Based on the receptor analysis, the high-metastatic cancer cells seemed to have abundant expression of collagen-binding (GPIa-IIa-like), fibrinogen-binding (GPIIb-IIIa-like) and sialoprotein-binding (P-selectin-like) antigens (Fig. 2B). Therefore the glass slide surfaces for these experiments were modified with fibrinogen-derived Arginine–Glycine Aspartic acid (RGD) containing peptide (called P1 from here on) with the sequence cyclo-CNPRGDY(OEt)RC,40 or sialoprotein-derived P-selectin binding peptide (called P2 from here on) with the sequence DAEWVDVS,41 or a collagen-mimetic peptide (called P3 from here on) with the sequence [GPO]7.42 All peptides were synthesized using fluoroenylmethyloxycarbonyl chloride (Fmoc)-based solid-phase chemistry on a Knorr resin and characterized using mass spectrometry. The flow of cells over the modified surfaces was maintained for 15 min and the binding of the cells on the surfaces was imaged (DAPI fluorescence) using the Zeiss inverted fluorescence microscope. The relative binding of the MCF-7 cells versus the MDA-MB-231 cells on these surfaces was used to validate the receptor analysis data and to choose the ligands suitable for fabrication of platelet-inspired nanoconstructs for metastatic cell targeting.
2.4. Fabrication of platelet-inspired liposomal nanovehicles
Based upon results of the experiments described in Section 2.3 (results shown in Fig. 2 and 3), all three peptides P1, P2 and P3 were found to be able to bind the high-metastatic MDA-MB-231 cells more than the low-metastatic MCF-7 cells (higher capture of MDA-MB-231 compared to MCF-7 on modified surfaces), with P1 and P2 showing statistical significance in this enhanced binding capability. Additionally, the collagen-binding interaction with the peptide P3 may have limited applicability in developing cancer cell-binding drug delivery systems, since the collagen interaction is relevant mostly at sites of endothelial injury and denudation at the vascular wall, and not so much in the circulating blood volume. Therefore, we focused on the two receptor systems (GPIIb-IIIa-like receptors and P-selectin-like receptors) and postulated that nanoconstructs bearing surface decoration of P1 and P2 peptides can have high degree of binding to the MDA-MB-231 cells compared to the MCF-7 cells. Additionally, it has been shown in several recent studies that vehicles binding simultaneously to multiple types of receptors on a target cell (heteromultivalent interactions) increases the targeted binding specificity and anchorage strength under a dynamic (e.g. circulation) environment.43 Rationalizing from such observations, using liposomes as model nanovehicles we developed platelet-inspired nanoconstructs bearing heteromultivalent surface-decoration with P1 and P2 peptides. For this, the peptides were conjugated via their amino (–NH2) termini to the carboxyl (–COOH) termini of the lipid DSPE-PEG2000-COOH via carbodiimide chemistry, resulting in DSPE-PEG-P1 and DSPE-PEG-P2 conjugates. Details of the conjugation methods and characterization of conjugates are available in our previous reports.37,45 These DSPE-PEG-peptide conjugates, at 2.5 mol% per conjugate type (i.e. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 peptide ratio), were combined with 50 mol% DSPC and 45 mol% cholesterol to fabricate heteromultivalently peptide-decorated liposomes via the reverse phase evaporation and extrusion (RPEE) technique.44 Liposome size was characterized by dynamic light scattering to reveal a mean value of 150 nm in diameter, similar to previously reported studies.45
:1 peptide ratio), were combined with 50 mol% DSPC and 45 mol% cholesterol to fabricate heteromultivalently peptide-decorated liposomes via the reverse phase evaporation and extrusion (RPEE) technique.44 Liposome size was characterized by dynamic light scattering to reveal a mean value of 150 nm in diameter, similar to previously reported studies.45
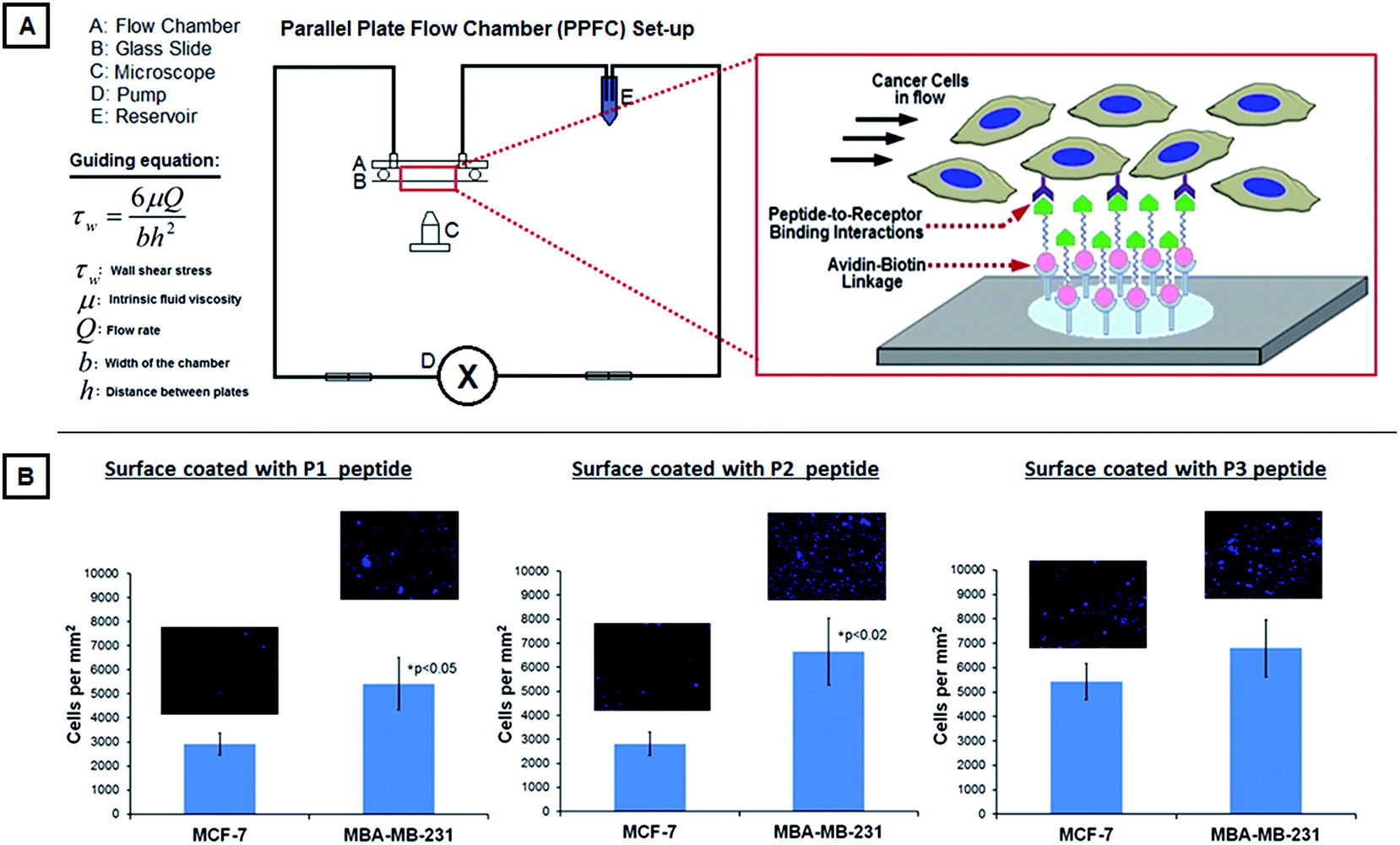 | ||
| Fig. 3 [A] shows the experimental PPFC set-up for studying the binding of DAPI-stained MCF-7 versus MDA-MB-231 cells on glass slides coated with P1, P2 or P3 peptides; the figure component also shows the working equation for the PPFC set-up by virtue of which the shear stress was maintained at 5 dyn cm−2 on the slide surface. [B] shows representative fluorescent images (DAPI-stained blue nuclei) along with analyzed quantitative data of cell binding on the peptide-modified surfaces, showing that all three peptides bind the high-metastatic MDA-MB-231 cells more than the low-metastatic MCF-7 cells, with P1 and P2 peptides showing statistical significance in this enhanced binding; these results support the receptor expression data (Table in Fig. 2) on the two cell types and also suggest that the peptides can be potentially used to design nanoconstructs actively targeted to the high-metastatic MDA-MB-231 cells. | ||
2.5. Doxorubicin loading and release studies with the platelet-inspired nanoconstructs
Doxorubicin–HCl (DOX) was loaded into the platelet-inspired nanoconstructs using transmembrance pH gradient method.46,47 For this, using 0.5 mol L−1 sodium bicarbonate, the external pH was adjusted to ∼11. Based on a 1:10 drug/lipid mole ratio, DOX solution was prepared at 0.58 mg mL−1 in 0.9% NaCl solution and pre-heated to 65 °C. This DOX solution was added to the liposome solution and allowed to incubate at 65 °C under mild stirring for 20 minutes. To determine resultant encapsulation efficiency, the DOX-encapsulated liposomal nanoconstructs were ultracentrifuged at 100000 rpm for 20 minutes to produce a liposome pellet, and unencapsulated DOX was removed by aspirating the supernatant. The lipid pellet was subsequently lysed with 0.1% triton-X in PBS, and DOX concentration from the lysed pellet as well as from the removed supernatant was quantified using UV-Visible spectrometry at 480 nm absorbance. Encapsulation efficiency was determined using the equation:where Cpellet and Csupernatant are DOX concentrations from the pellet and supernatant respectively, as estimated by DOX absorbance calibration curves. In order to determine the release kinetics of DOX from the PILs, in separate experiments, the pellet formed from ultracentrifugation of DOX-loaded nanoconstructs was resuspended in 1 mL PBS and transferred into 1000 MWCO dialysis tubing. The tubings were sealed and the suspension was allowed to dialyze in a beaker containing 50 mL PBS with stirring at 37 °C. At various time points, 200 μL of dialysate was removed and analyzed using UV spectrometry at 480 nm absorbance to determine released DOX concentration as estimated by corresponding calibration curves. The UV spectroscopy data was additionally validated by fluorescence spectrometry measurements with 501 nm excitation/555 nm emission. Dialysate removed for such DOX release quantification was replaced with equal volumes of 1X PBS.

2.6. Targeted binding and drug delivery in MDA-MB-231 versus MCF-7 cell monocultures
MCF-7 and MDA-MB-231 cells were seeded into 96 well plates at 10000 cells per well. The seeded wells were treated with DOX-loaded non-targeted liposomes in media (9 μg total DOX per mL), DOX-loaded platelet-inspired liposomal nanoconstructs in media (9 μg total DOX per mL), free DOX in media (9 μg total DOX per mL), or plain media without any DOX (untreated). Cells were allowed to incubate with treatments for 1, 3 and 5 hours at 37 °C under gentle shaking at 100 rpm. The incubation solutions were subsequently removed, and the cells were washed gently with DPBS. Media was then added to the washed cells, and cells were allowed to grow for another 48 h under normal culture conditions. After 48 hours, a standard MTT assay was used to assess metabolic activity (hence viability) of the cells. The MTT-based metabolic activity was quantified with UV-visible spectrometry, measuring absorbance at 540 nm. In order to evaluate the delivery of DOX (with natural orange-red fluorescence) to the cells by the DOX-loaded platelet-inspired nanoconstructs compared to the DOX-loaded non-targeted liposomes, fluorescent images were taken. For this, the cells were stained with Cell Trace™ CFSE (green fluorescence, λex = 488 nm, λem = 521 nm) following the manufacturer's protocol. After treatment with DOX-loaded platelet-inspired nanoconstructs or DOX-loaded non-targeted liposomes for 5 hours, followed by removal of liposome suspensions and subsequent 48 hour incubation with media at 37 °C, the cells were fixed with 4% paraformaldehyde for 15 minutes and imaged using the Zeiss inverted fluorescence microscope to obtain multi-colored (green of CFSE and orange-red of DOX) images using appropriate filters. The co-localization of green and orange-red was considered to be a visual confirmation of DOX presence within the cells.
2.7. Targeted drug delivery studies to MCF-7 and MDA-MB-231 cells in co-cultures
The monoculture studies described in Section 2.6 were aimed at establishing whether the platelet-inspired liposomal nanoconstructs were capable of enhanced targeting and DOX delivery to the high-metastatic cells, compared to the low-metastatic cells. In further advancement of the studies, we aimed at investigating whether this cell-specific targeted binding and DOX delivery to the high-metastatic cell lines can happen in the presence of other cells that do not abundantly express the platelet-relevant receptors. For ‘proof-of-concept’, we rationalized that since MCF-7 cells do not highly express the platelet-relevant receptors, they can in essence become ‘control cells’ in co-culture with the MDA-MB-231 cells since the latter express the platelet-relevant receptors abundantly. Therefore for the co-culture studies, the MCF-7 cells were pre-stained green with Cell Trace™ CFSE and MDA-MB-231 were pre-stained violet with Cell Trace™ Violet, and cells were seeded into the same wells of a 12 well plate at densities of 15000 cells per well per cell type. Cells were allowed to grow in normal culture conditions over night. The resultant co-cultures were incubated with DOX-loaded non-targeted liposome suspension or DOX-loaded platelet-inspired liposomal nanoconstruct suspension (added to media at 9 ug mL−1) for 4 h. The liposome-containing media was then removed, fresh media was added and the co-cultures were further maintained at 37 °C in the incubator for 48 hours. Following this, the cells were gently washed with DPBS and fixed with 4% PFA for 15 minutes. For visual analysis of preferential DOX delivery in co-culture, the cells were imaged with the inverted fluorescence microscope to capture images of DOX (orange-red) co-localization with green (MCF-7) and violet (MDA-MB-231) cells. Also, for multiple batches of such experiments (n = 15), the number of residual fixed green cells and violet cells were counted in each image using Image J (NIH) and the corresponding cell counts were considered to be reflective of ‘still viable’ cells. This consideration is based on the rationale that cells that had died due to DOX action had already lifted off and consequently been removed during the washing phase, leaving only still viable cells in the wells to be subsequently fixed and imaged. Furthermore, according to the American Type Culture Collection (ATCC) propagation protocols for MDA-MB-231 and MCF-7 breast adenocarcinoma cell lines, population doubling times (PDT) for both cell lines are approximately 38 hours. Therefore we rationalized that any significant differences in post-treatment population viability can be attributed to preferential delivery of DOX to one cell line over the other. Therefore, this assay provided a rational way to report comparative cell viability in co-culture conditions.
2.8. Statistical analysis
Student's t-test was used to analyze the difference between two means. All other statistical analyses between multiple groups were performed using one-way ANOVA with Tukey method. In all analyses, significance was considered to be p < 0.05.3. Results
3.1. Interaction of cancer cells with human platelets and analysis of relevant antigen expression on cancer cells
Fig. 2A shows representative multispectral fluorescence images of calcein-stained (green) active platelets bound to phalloidin (red) and DAPI (blue) co-stained MDA-MB-231 versus MCF-7 cells. It is to be noted that there is a morphological and dimensional difference between the two cell lines, at the same scale of magnification. As evident from the images, active platelets have a significantly higher extent of binding to the high-metastatic MDA-MB-231 cells compared to the low-metastatic MCF-7 cells. The results shown in Fig. 2A can be explained by the receptor (antigen) expression data shown in Fig. 2B, where the highest levels of expression on MDA-MB-231 cells compared to MCF-7 cells were for GPIa-IIa-like antigens (collagen-binding capability), GPIIb-IIa-like integrins (fibrinogen-mediated platelet-binding capability) and P-selectin-like receptors (sialoprotein-mediated active platelet-binding capability). The binding mechanisms were further validated by the cell-binding experiments in flow over the ligand-modified surfaces. Fig. 3 shows the results from these experiments with surfaces modified by GPIIb-IIIa-binding P1 peptide ligands, P-selectin binding P2 peptide ligands and GPIa-IIa-binding P3 peptides. As evident from the results, all three surfaces bind high-metastatic MDA-MB-231 cells more than the low-metastatic MCF-7 cells, with P1 and P2-modified surfaces showing statistical significance in the enhanced binding. The rationale for limited utility of the collagen-mimetic peptide (P3) for metastatic cell-targeting was stated previously in Section 2.4. Therefore, from a targeted drug delivery standpoint, liposome-based platelet-inspired nanoconstructs were developed by heteromultivalently decorating the liposome surface (at 1:1 ratio) with 2.5 mol% each of P1 peptide and P2 peptide. These nanoconstructs were loaded with DOX and their capability of cell-specific binding, DOX delivery and preferential killing of MDA-MB-231 versus MCF-7 cells were studied.
3.2. Doxorubicin loading in platelet-inspired constructs and release kinetics characterization
The model cancer drug DOX was loaded into the platelet-inspired liposomal nanoconstructs via established pH gradient method. Fig. 4A and B show the DOX absorbance and fluorescence calibration data, based on which the encapsulation efficiency (EE) was determined. Fig. 4C shows four batches of EE analysis. As evident from the data, the average EE was ∼90%. It is to be noted that for the purpose of proof-of-concept in vitro experiments the average actual amount of drug loaded was ∼0.5 mg mL−1, which is lower than the clinically relevant in vivo liposomal DOX formulation of Doxil® (2 mg mL−1). The DOX-loaded liposomal nanoconstructs were subjected to incubation in PBS at physiological pH 7.4 versus tumor extracellular pH 6.5. Fig. 4D shows the DOX release profile in these conditions. As evident from the data, the release followed first order kinetics in both release conditions, and the more acidic pH resulted in a slightly higher % release over time. Future refinement of the nanoconstruct membrane composition with more acid-labile (i.e. degradable at lower pH) components will be geared towards accelerating the pH-triggered enhanced release of DOX in the tumor extracellular (pH ∼ 6.5), as well as intracellular lysosomal (pH ∼ 3–4) environment. | ||
| Fig. 4 [A] and [B] show respectively the DOX absorbance and fluorescence calibration curves based on which the encapsulation efficiency (EE) of DOX in the PILs was determined. [C] is the EE analysis from four batches of PILs, showing that the average EE was ∼90%. [D] shows the DOX release profile from the PILs in two pH conditions, physiological (pH 7.4) and tumor extracellular (pH 6.5), demonstrating that the DOX gets released via first order kinetics. | ||
3.3. Targeted DOX delivery and cell killing using the platelet-inspired nanovehicles in cancer cell monocultures
Following characterization of loading and release of DOX from the platelet-inspired liposomal (PIL) nanoconstructs, the resultant DOX-loaded formulations were tested for their cell targeting and killing capability on monocultures of the low-metastatic MCF-7 versus the high-metastatic MDA-MB-231 human breast cancer cells. As described in the Methods section, the experimental conditions involved (a) no treatment, (b) treatment with DOX-loaded non-targeted liposomes, (c) treatment with DOX-loaded platelet-inspired nanoconstructs, and (d) free DOX, and the resultant DOX-induced cell killing was evaluated by standard MTT assay. Fig. 5A shows representative fluorescent images (at the 5 h incubation period) of Cell Trace™ CFSE-stained green fluorescent MCF-7 or MDA-MB-231 cells in monoculture incubated with DOX-loaded (orange-red fluorescence) non-targeted liposomes versus DOX-loaded PIL nanoconstructs. As evident from the images, the platelet-inspired vehicles resulted in significantly enhanced delivery of DOX to the MDA-MB-231 cells, indicated by the increased co-localization of the orange-red DOX fluorescence with the green cell fluorescence in Fig. 5A(iv). Fig. 5B shows the cell viability results for 1 h, 3 h and 5 h treatments, as analyzed by post-treatment MTT. As evident from the data, compared to ‘no treatment’ group and ‘incubated with DOX-loaded non-targeted liposome’ group, the ‘incubated with DOX-loaded platelet-inspired liposome group’ showed significantly lower viability (i.e. higher killing) of MDA-MB-231 cells over time, especially at 5 h treatment time (*). Also, during this 1–5 h treatment period, although some initial killing was seen in the MCF-7 cells incubated with the DOX-loaded platelet-inspired vehicles, the extent of killing did not increase with time of incubation. This suggests that because of in vitro pooled incubation condition, there is some extent of non-specific nanovehicle uptake in both types of cells resulting in some initial extent of killing, but the DOX delivery is enhanced in the MDA-MB-231 cells by the platelet-inspired vehicles over time, which resulted in DOX-induced increased cytotoxic effect in these cells over time compared to the MCF-7 cells. It is to be noted that compared to both non-targeted and targeted formulations, free DOX resulted in higher cytotoxicity at all treatment times. This is possibly due to higher membrane-mediated direct uptake of DOX in pooled incubation conditions, but from a clinical perspective free DOX administration has very limited applicability because of its systemic toxicity risks.48 Also, in a realistic delivery situation free DOX will not be present in a static pooled condition proximal to cells for fast intracellular uptake. In this context, cell surface-bound DOX-loaded platelet-inspired nanovehicles can have high capability to deliver DOX and kill metastatic cells. It is also to be noted that in the in vitro experimental conditions the cell-killing extent is only moderate (about 40–50%). This is possibly because our drug dose is significantly lower (∼0.5 mg mL−1) compared to the clinically used Doxil® dose (2 mg mL−1). The focus of the current studies is to demonstrate that the platelet-inspired nanovehicles can deliver DOX in a targeted way to metastatic cells to render higher killing of these cells compared to non-metastatic cells. Future studies will be focused on optimizing the DOX dosing in these nanovehicles to maximize the metastatic cell-killing extent. | ||
| Fig. 5 [A] shows representative fluorescent images (at the 5 h incubation period) of Cell TraceTM CFSE-stained green fluorescent MCF-7 versus MDA-MB-231 cells in monoculture incubated with DOX-loaded (orange-red fluorescence) non-targeted liposomes versus DOX-loaded targeted platelet-inspired liposome (PIL) nanoconstructs; the targeted PILs resulted in significantly enhanced delivery of DOX to the MDA-MB-231 cells compared to the MCF-7 cells, indicated by the increased co-localization of the orange-red DOX fluorescence with the green cell fluorescence (scale bar represents 100 μm). [B] shows the cell viability data of MCF-7 cells versus MDA-MB-231 cells treated with DOX-loaded PILs, along with relevant control formulations; as evident from the data, compared to ‘no treatment’ group and ‘incubated with DOX-loaded non-targeted liposome’ group, incubation with DOX-loaded ‘targeted PIL’ showed significantly lower viability (i.e. higher killing) of MDA-MB-231 cells, especially after the 5 h treatment time (‘*’ represents p < 0.005). Also, at this treatment time, there was no statistical increase of killing of the MCF-7 cells incubated with DOX-loaded targeted PILs. The ‘free DOX’ treatment resulted in even higher extent of killing (‘#’ represents p < 0.005 when compared to the ‘targeted PIL’ group), but free DOX administration has clinical limitations due to significant systemic toxicity issues. | ||
3.4. Cell-specific DOX-delivery and cell-killing by the platelet-inspired nanoconstructs incubated in cancer cell co-cultures
As explained in the Methods section, the MDA-MB-231 were stained violet, the MCF-7 cells were stained green, and the DOX has inherent orange-red fluorescence. Fig. 6A shows representative multi-spectral fluorescence images of cell co-cultures (violet MDA-MB-231 + green MCF-7) incubated with the DOX-loaded non-targeted liposomes versus DOX-loaded platelet-inspired nanoconstruct vehicles. The non-targeted liposomes had only minimal capability to deliver DOX to either cell lines in co-culture, as indicated by minimal co-localization of DOX red fluorescence with either the violet (MDA-MB-231) or green (MCF-7) fluorescent cells. In contrast, incubation with the platelet-inspired vehicles indicated preferential enhanced delivery of DOX to the high-metastatic MDA-MB-231 cells compared to low-metastatic MCF-7 cells, as evident from the increased co-localization of the DOX orange-red fluorescence with the violet MDA-MB-231 cells compared to that with the green MCF-7 cells. Fig. 6B shows the resultant cell-viability data from these studies, estimated by the assay described in the Methods sections. The data suggests that even in co-culture conditions the increased preferential delivery of DOX to the MDA-MB-231 cells by the platelet-inspired nanovehicles resulted in enhanced killing of these cells compared to MCF-7 cells. The non-targeted liposomes showed only minimal killing efficacy towards either cell line, possible due to some non-specific uptake resulting from pooled incubation conditions. | ||
| Fig. 6 [A] shows representative multi-spectral fluorescence images of cell co-cultures (CellTrace™ Violet-stained blue-violet MDA-MB-231 + CellTrace™ CFSE-stained green MCF-7) incubated with the DOX-loaded non-targeted liposomes versus DOX-loaded targeted platelet-inspired liposome (PIL) nanoconstructs. Minimal co-localization of DOX orange-red fluorescence with either the violet (MDA-MB-231) or green (MCF-7) fluorescent cells in [A]i indicates that non-targeted liposomes had minimal capability to deliver DOX to either cell lines in co-culture, while increased co-localization of the DOX orange-red fluorescence with the violet MDA-MB-231 cells compared to green MCF-7 cells (scale bar represents 100 μm), [A]ii indicates that incubation with the targeted PILs resulted in preferential enhanced delivery of DOX to the pro-metastatic MDA-MB-231 cells compared to low metastatic MCF-7 cells. [B] shows the resultant cell-viability data, indicating that the increased preferential delivery of DOX to the MDA-MB-231 cells by the targeted PILs resulted in enhanced killing of these cells compared to MCF-7 cells, in co-culture, while the non-targeted liposomes showed only minimal killing efficacy towards either cell lines. | ||
4. Discussion
Detection, treatment and prevention of metastasis are crucial in reducing malignancy-related mortalities. However, due to the heterogeneity in size, location, target antigen expression and signaling pathways, metastasis poses a formidable challenge in cancer therapies. Current clinical strategies to treat metastasis principally rely on systemic administration of chemotherapeutics, immunotherapeutics and signal-inhibiting biologic small molecules, which can work effectively if they can sufficiently localize at the metastatic sites but otherwise, can pose severe systemic side-effects due to indiscriminate action on non-target healthy tissues. To ensure localization of these agents to metastatic sites, one promising strategy is the use of drug delivery systems like micro- and nanoparticles that can encapsulate the agents and bind to metastatic cells or sites for selective delivery. This strategy is being utilized to develop actively targeted drug delivery vehicles that bind to receptors like Epidermal Growth Factor Receptors (EGFR), Folate Receptors (FR), Transferrin Receptor (TfR), integrin αVβ3 etc. that have been reported to be significantly upregulated in growing tumors.49–53 However, most of these targetable receptors were identified from primary tumors, and there is significant clinical data that the expression levels of these antigens can be highly heterogeneous at metastatic locations.18–21 This suggests that utilizing such receptors for site-specific or cell-specific binding of drug delivery vehicles may not always be effective for metastasis. Based on this rationale, we are investigating alternative strategies of specific binding to metastatic cells and one such strategy is to utilize the binding interactions between metastatic cells with cells of the vascular compartment, especially platelets. The role of platelets in facilitating hematologic metastasis via direct molecular binding interactions with pro-metastatic cells as well as via local action of platelet secretome components have been suggested by several studies.28–36 These reports provide a rationale to consider platelet-cancer cell interactions as a paradigm to engineer drug delivery systems that utilize similar interactions to bind to metastatic cells for preferential delivery of anti-cancer agents.Based on the above rationale, we have studied the interaction of ADP-activated platelets with high-metastatic (MDA-MB-231) versus low-metastatic (MCF-7) human breast cancer cells, in vitro in a flow environment. Our studies established that the active platelets have high interaction capability with the MDA-MB-231 cells compared to the MCF-7 cells. Our subsequent immunofluorescent studies have identified several possible ligand–receptor mechanisms that can allow such interactions, and our microfluidic assays validated the utilization of these interactions for binding to high-metastatic cells. Consequently, we have utilized two of these interaction mechanisms simultaneously on a model liposomal platform by heteromultivalently decorating liposomes with two peptides, P1 binding to GPIIb-IIIa-like integrins and P2 binding to P-selectins. The resultant platelet-inspired liposomal nanovehicles showed enhanced binding and DOX delivery to the MDA-MB-231 cells compared to MCF-7 cells in monoculture, resulting in significant killing of the MDA-MB-231 cells. While monoculture based studies establish the enhanced binding and DOX-delivery of the platelet-inspired vehicles in high-metastatic cell type versus low-metastatic cell type, it is not sufficient to establish the extent of preferential delivery of DOX to MDA-MB-231 cells in presence of another non-metastatic cell type. Therefore, further studies were carried out where the DOX-loaded platelet-inspired vehicles were incubated with MDA-MB-231 cells and MCF-7 cells in co-culture, with the rationale that the MCF-7 cells, because of the lack of platelet-interactive receptor antigens on them, would act as representative ‘negative control’ cells in the co-culture. These co-culture experiments established that the platelet-inspired interactions by the liposomal vehicles not only enhanced the preferential binding and resultant DOX delivery to MDA-MB-231 even in presence of representative ‘non-target’ control cells (in this case low-metastatic MCF-7 cells).
It is to be noted that the studies reported here were carried out using liposomes as a model drug delivery platform, MDA-MB-2321 cells as model high-metastatic cell line, DOX as a model anti-cancer drug and a total ligand incorporation of 5 mol% (i.e., 2.5. mol% each of P1 and P2 peptides) for proof of concept. For further advancement this approach, future research may include fine-tuning the delivery vehicle platform itself (e.g. using stimuli-sensitive lipidic or polymeric particles to modulate drug release kinetics), as well as, optimizing the ligand decoration of the vehicles (e.g. modulating surface-density and chemistry of ligands to maximize target cell binding affinity and selectivity). Also a variety drugs and imaging probes can become candidate payloads for metastasis-targeted delivery, and the platelet-inspired targeting strategy can be subsequently studied across a variety of pro-metastatic cancer cell lines to evaluate the broad-spectrum applicability of this platelet-inspired technology. The ultimate test of this technology will be to evaluate its ability to become a ‘seek and destroy’ system that can potentially bind to circulating metastatic cells, as well as, micrometastatic foci in vivo to deliver anti-tumor drugs. Future research will be directed towards evaluating this ability in suitable in vivo models of metastatic malignancies.
5. Conclusion
The current study involves utilization of platelet interactions with pro-metastatic cancer cells to develop platelet-inspired nanovehicles for targeted delivery of anti-cancer agents to metastasis. To this end, platelet-inspired liposomal systems were developed that can simultaneously bind to GPIIb-IIIa-like integrins and P-selectins on the high-metastatic MDA-MB-231 human breast cancer cells. Loading of DOX within these nanovehicles was achieved using a pH-gradient based method and the release of DOX from the construct vehicles showed first order kinetics. The platelet-inspired mechanisms on the nanovehicles rendered enhanced binding and delivery of DOX to MDA-MB-231 cells in monoculture, as well as, in co-culture with the non-target MCF-7 cells. This enhanced delivery resulted in higher killing of the MDA-MB-231 cells, both in monoculture and co-culture set-up. Our results suggest the promise of such platelet-inspired approach in designing nanomedicine platforms for metastasis-targeted drug delivery.Acknowledgements
The research was supported by American Cancer Society (ACS) Institutional Research Grant (# IRG-91-022-18, PI: Sen Gupta) at Case Western Reserve University. Modery-Pawlowski is supported by NSF GRFP Grant Number DGE-0951783. Preethi Siva was partly supported by Provost Summer Undergraduate Research Grant (P-SURG) at Case Western Reserve University.References
- P. Mehlen and A. Puisieu, Nat. Rev. Cancer, 2006, 6, 449–458 CrossRef CAS PubMed.
- C. Bernard-Marty, F. Cardoso and M. Piccart, Oncologist, 2004, 9, 617–632 CrossRef PubMed.
- Metastatic Cancer Fact Sheet – National Cancer Institute, 2014.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS PubMed.
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, J. Controlled Release, 2012, 161, 175–187 CrossRef CAS PubMed.
- E. K. H. Chow and D. Ho, Sci. Transl. Med., 2013, 5, 216rv4 CrossRef PubMed.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958–962 CrossRef CAS PubMed.
- C. M. Dawidczyk, C. Kima, J. H. Park, L. M. Russell, K. H. Lee, M. G. Pomper and P. C. Searson, J. Controlled Release, 2014, 187, 133–144 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2011, 53, 282–318 Search PubMed.
- K. F. Pirollo and E. H. Chang, Trends Biotechnol., 2008, 26, 552–558 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- A. Lluch, I. Álvarez, M. Muñoz, M. A. Seguí, I. Tusquets and L. García-Estévez, Crit. Rev. Oncol. Hematol., 2014, 89, 62–72 CrossRef PubMed.
- A. Schroeder, D. A. Heller, M. M. Winslow, J. E. Dahlman, G. W. Pratt, R. Langer and D. G. Anderson, Nat. Rev. Cancer, 2012, 12, 39–50 CrossRef CAS PubMed.
- G. Mattheolabakis, B. Rigas and P. P. Constantinides, Nanomedicine, 2012, 7, 1577–1590 CrossRef CAS PubMed.
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevick-Muraca, W. Zamboni, O. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS PubMed.
- J. W. Nichols and Y. H. Bae, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS PubMed.
- P. S. Steeg, Clin. Cancer Res., 2008, 14, 3643–3645 CrossRef PubMed.
- N. Niikura, J. Liu, N. Hayashi, E. A. Mittendorf, Y. Gong, S. L. Palla, Y. Tokuda, A. M. Gonzalez-Angulo, G. N. Hortobagyi and N. T. Ueno, J. Clin. Oncol., 2012, 30, 593–599 CrossRef PubMed.
- T. Kuwai, T. Nakamura, S. J. Kim, T. Sasaki, Y. Kitadai, R. R. Langley, D. Fan, S. R. Hamilton and I. J. Fidler, Am. J. Pathol., 2008, 172, 358–366 CrossRef PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- H. Esmaeilsabzali, T. V. Beischlag, M. E. Cox, A. M. Parameswaran and E. J. Park, Biotechnol. Adv., 2013, 31, 1063–1084 CrossRef CAS PubMed.
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, J. Cell Biol., 2011, 192, 373–382 CrossRef CAS PubMed.
- C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559–1564 CrossRef CAS PubMed.
- I. Baccelli and A. Trumpp, J. Cell Biol., 2012, 198, 281–293 CrossRef CAS PubMed.
- M. Książkiewicz, A. Markiewicz and A. J. Żaczek, Pathobiology, 2012, 79, 195–208 CrossRef PubMed.
- R. Kalluri and R. A. Weinberg, J. Clin. Invest., 2009, 119, 1420–1428 CrossRef CAS PubMed.
- P. Mehta, Blood, 1984, 63, 55–63 CAS.
- B. Felding-Habermann, T. E. O'Toole, J. W. Smith, E. Fransvea, Z. M. Ruggeri, M. H. Ginsberg, P. E. Hughes, N. Pampori, S. J. Shattil, A. Saven and B. M. Mueller, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1853–1858 CrossRef CAS PubMed.
- K. Lawler, G. Meade, G. O'Sullivan and D. Kenny, Am. J. Physiol.: Cell Physiol., 2004, 287, C1320–C1327 CrossRef CAS PubMed.
- H. Läubli and L. Borsig, Semin. Cancer Biol., 2010, 20, 169–177 CrossRef PubMed.
- J. S. Palumbo, K. E. Talmage, J. V. Massari, C. M. La Jeunesse, M. J. Flick, K. W. Kombrinck, M. Jirousková and J. L. Degen, Blood, 2005, 105, 178–185 CrossRef CAS PubMed.
- M. Labelle, S. Begum and R. O. Hynes, Cancer Cell, 2011, 20, 576–590 CrossRef CAS PubMed.
- L. J. Gay and B. Felding-Habermann, Nat. Rev. Cancer, 2011, 11, 123–134 CrossRef CAS PubMed.
- D. Schumacher, B. Strilic, K. K. Sivaraj, N. Wettschureck and S. Offermanns, Cancer Cell, 2013, 24, 130–137 CrossRef CAS PubMed.
- N. M. Bambace and C. E. Holmes, J. Thromb. Haemostasis, 2011, 9, 237–249 CrossRef CAS PubMed.
- C. L. Modery-Pawlowski, A. M. Master, V. Pan, G. P. Howard and A. Sen Gupta, Biomacromolecules, 2013, 14, 910–919 CrossRef CAS PubMed.
- G. B. Fields and R. L. Noble, Int. J. Pept. Protein Res., 1990, 35, 161–214 CrossRef CAS PubMed.
- Y. Geng, S. Chandrasekaran, J. W. Hsu, J. M. Gidwani, A. D. Hughes and M. R. King, PLoS One, 2013, 8, e54959 CAS.
- C. P. Cheng, D. Parker and C. A. Taylor, Ann. Biomed. Eng., 2002, 30, 1020–1032 CrossRef.
- S. Cheng, W. S. Craig, D. Mullen, J. F. Tschopp, D. Dixon and M. D. Pierschbacher, J. Med. Chem., 1994, 37, 1–8 CrossRef CAS.
- C. C. Appeldoorn, T. J. M. Molenaar, A. Bonnefoy, S. H. Van Leeuwen, P. A. H. Vandervoort, M. F. Hoylaerts, T. J. Van Berkel and E. A. Biessen, J. Biol. Chem., 2003, 278, 10201–10207 CrossRef CAS PubMed.
- X. Mo, Y. An, C. S. Yun and S. M. Yu, Angew. Chem., Int. Ed., 2006, 45, 2267–2270 CrossRef CAS PubMed.
- C. L. Modery-Pawlowski and A. Sen Gupta, Biomaterials, 2014, 35, 2568–2579 CrossRef CAS PubMed.
- C. L. Modery, M. Ravikumar, T. L. Wong, M. J. Dzuricky, N. Durongkaveroj and A. Sen Gupta, Biomaterials, 2011, 32, 9504–9514 CrossRef CAS PubMed.
- A. Wagner and K. Vorauer-Uhl, J. Drug Delivery, 2011, 591325 Search PubMed.
- L. D. Mayer, M. B. Bally and P. R. Cullis, J. Liposome Res., 1990, 1, 463–480 CrossRef.
- G. Niu, B. Cogburn and J. Hughes, Methods Mol. Biol., 2010, 624, 211–219 CAS.
- L. M. Kaminska, V. M. McLeod, B. D. Kelly, G. Sberna, B. J. Boyd, M. Williamson, D. J. Owen and C. J. H. Porter, Nanomedicine, 2012, 8, 103–111 CrossRef PubMed.
- A. M. Master and A. Sen Gupta, Nanomedicine, 2012, 7, 1895–1906 CrossRef CAS PubMed.
- J. M. Qian, H. Li, H. Sun and K. Ho, Pharmacol. Rev., 2002, 54, 561–587 CrossRef.
- S. A. Kularatne and P. S. Low, Methods Mol. Biol., 2010, 624, 249–265 CAS.
- Z. Liu, F. Wang and X. Chen, Drug Dev. Res., 2008, 69, 329–339 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |