
From nanorods of palygorskite to nanosheets of smectite via a one-step hydrothermal process†
Abstract
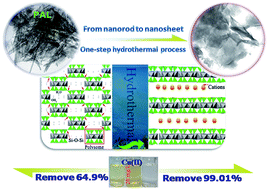
The structural evolution of silicate opens a new avenue to cognize its microstructure, intensify its properties and extend its application. Herein, the one-step transformation of palygorskite (PAL) nanorods into smectite nanosheets was successfully achieved under mild hydrothermal condition with no addition of any extra chemicals. The structural evolution of PAL at different reaction stages and the change in physico-chemical characteristics was intensively studied through field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), thermogravimetry (TG) and Fourier transform infrared spectroscopy (FTIR) techniques. The key factors determining the transformation process were clarified. It was found that the moderate mechanical grinding, pH values and the existence of dolomite are essential to realize the transformation, and alkaline condition may facilitate the transformation. The transformation from nanorods to nanosheets is a rebuilding process of crystals, and the c/2 slide of tetrahedrons represents the main transformation mechanism. After the hydrothermal process, the adsorption capability of RPAL for Cu(II) evidently enhanced by 167%, and 99.01% of Cu(II) ions (only 64.9% for raw PAL) can be removed from 20 mg L−1 of Cu(II) solution.
 Please wait while we load your content...
Please wait while we load your content...

