DOI:
10.1039/C5RA05073A
(Paper)
RSC Adv., 2015,
5, 50747-50755
Synthesis of chlorostannate(II) ionic liquids and their novel application in the preparation of high-quality L-lactide†
Received
22nd March 2015
, Accepted 2nd June 2015
First published on 2nd June 2015
Abstract
Polylactic acid (PLA) is a representative biodegradable polymer, which is expected to be a promising replacement for some petroleum-based materials. Noticeably, the properties of PLA products depend strongly on the quality of the lactide monomer, a crucial precursor of PLA production. In this work, a large range of different chlorostannate(II) ionic liquids (ILs), prepared by mixing 1-butyl-3-methyl-imidazolium chloride and tin(II) chloride in various molar ratios, xSnCl2, were firstly applied for the preparation of L-lactide of high chemical and optical purity. The cation–anion interaction, the thermal stability and the acidity of imidazolium-based chlorostannate(II) ionic liquids were experimentally determined and systematically analyzed. Compared with the conventional SnCl2 catalyst, the depolymerization of oligomeric poly(L-lactic acid) catalyzed by chlorostannate(II) ionic liquids occurred in a moderate yield. Interestingly, using [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) as a catalyst, L-lactide of 99.9% optical purity was obtained, simultaneously leaving a high-Mw oligomeric residue with high isotacticity (99.1%). Furthermore, the effects of various reaction parameters were investigated in order to obtain the highest possible yield of lactide. A plausible reaction mechanism was suggested and discussed. Finally, owing to the reutilization of PLLA residue of high isotacticity, a reiterative lactide synthesis was realized. The recycled catalyst showed no notable loss of activity. By combining this chlorostannate(II)-based IL (xSnCl2 = 0.63) catalyst technology with the cyclic resynthesis process, high-quality L-lactide could be selectively produced in high yield (>80%, based on L-lactic acid replenished).
Introduction
Owing to the solid waste disposal problems caused by petroleum-based plastics, there has been a sustained research interest in the development of biodegradable polymers as replacements over the past decade.1,2 Polylactic acid (PLA) is the front runner in the emerging bioplastics market with the best availability and the most attractive cost structure.3 PLA can be produced by condensation polymerization directly from its basic building block lactic acid, which is derived from sustainable sources.4 However, it was difficult to obtain a high molecular-weight (Mw) PLA using the conventional, direct condensation reaction, owing to the existence of the dehydrated equilibrium or the resulting depolymerization reaction.5 Currently, the industrially acceptable PLA synthetic process is the ring-opening polymerization (ROP) of lactide, the six-membered dimeric cyclic esters of lactic acid.6,7 In this process, the lactic acid monomer is first polymerized to low-Mw PLA, depolymerized or dimerized to lactide, and then re-polymerized to obtain high-Mw PLA.8 This mechanism does not generate additional water, and hence a wide range of molecular weights is accessible.9 It is found that processing, crystallization and degradation behavior of PLA all depend strongly on the structure and composition of the polymer chains.10 Isotactic poly(L-lactic acid) (PLLA) has high melting point, high crystallinity, and then better mechanical properties, such as higher modulus, than poly(D,L-lactic acid) (PDLLA).3,11 Thus, PLLA is usually used for an alternative to the petroleum-based polyester terephthalate for the preparation of films, fibers, and other plastic materials.12 L-Lactide is an important intermediate in the industrial production of high molar mass PLLA, and the quality of L-lactide is a crucial parameter for controlling the properties of the resulting PLLA products. Obviously, it is necessary that L-lactide should possess a high chemical and optical purity. Therefore, the synthesis of enantiomerically pure L-lactide is of practical importance for the preparation of high-Mw PLLA.
L-Lactide can be obtained by the thermal degradation of oligomeric PLLA (O-PLLA).13 A series of Sn, Al, Ti, Zn and Zr compounds have been used as catalysts for producing lactide from PLA oligomer.14,15 This process inevitably undergoes racemization because of an amply high temperature and an amply long reaction time.16 Yoo et al.17 demonstrated that the racemization of lactide occured via deprotonation due to its high sensitivity to weak bases, and this process was accelerated by increased temperature. Idage et al.9 synthesized L-lactide with a yield of about 98% using zinc and tin metal catalysts of less than 150 micron particle size, and the crude L-lactide was further purified through an extremely complicated process. The purification step mainly consisted of recrystallization of crude lactide from boiling anhydrous toluene, filtration and repeatedly washing of lactide crystals. Ehsani and co-workers18 carried out a comprehensive study of the lactide synthesis optimization. They showed that increasing temperature resulted in higher amounts of impurities in the crude lactide. Huang et al.19 successfully conducted the green synthesis of the enantiomerically pure L-lactide and D-lactide using the creatinine-guanidinium catalyst. Synthesis of L-lactide using an onium salt catalyst was reported by Ishijima et al.20 More various catalyst systems have been investigated to maximize the yield in synthesis of lactide, while minimizing the racemization and simplifying the purification. However, few studies have been focused on the ionic liquids (ILs) catalyst that can activate PLA oligomer.
Recently, halometallate ionic liquids (ILs) are frequently introduced as relatively clean and promising catalysts and solvents, because of their unique properties such as high thermal stability, potential recoverability and fine control over physical and chemical properties by wide selection of the halometallate anions.21–23 In particular, many studies on polymerization progresses catalyzed by ILs have been done with high catalytic activities.24–26 Nevertheless, little attention has been paid to the study of the synthesis of lactide from PLA oligomer using ILs as catalysts. Kim et al.27 prepared lactide using IL only as a solvent, resulting in a lower reaction temperature and higher mobility of the reactant. In our attempt to search for catalysts used in the depolymerization of O-PLLA, we speculate that chlorostannate(II) ionic liquids may be good candidates for three reasons. Firstly, the chlorostannate(II) ionic liquid can behave as a Lewis acid21 and some Lewis acids are catalysts for O-PLLA depolymerization. Secondly, the depolymerization of O-PLLA into L-lactide is normally carried out under high temperature and low pressure, which requires catalysts with high thermal stabilities. Chlorometallate ionic liquids are generally thermally stable and less volatile,28 which means that they may be excellent catalysts for depolymerization. Thirdly, chlorostannate(II) ionic liquids are more inexpensive than those incorporating gallium(III) or indium(III) and less sensitive toward moisture than those based on aluminum(III) and gallium(III).29 In this study, the imidazolium-based ionic liquids incorporating stannum(II) were successfully prepared. The structure, thermal stability, and acidity of these synthesized ILs were experimentally determined and systematically analyzed. Then the depolymerization of O-PLLA was carried out using these ILs as catalysts for the first time, to produce L-lactide with high optical purity. A possible catalysis mechanism was proposed. We also investigated the effects of various reaction parameters, such as temperature, reaction time, pressure and mole ratio of catalyst/O-PPLA. Furthermore, the residue polymers of depolymerization were reutilized for the resynthesis of L-lactide.
Experimental section
Materials
L-Lactic acid (L-LA) as a 90 wt% aqueous solution was purchased from Musashino Chemical (China) Company. Tin(II) chloride (Aladdin, water-free for synthesis, ≥99.0%), n-methylimidazole (Aladdin, ≥99.0%) and pyridine (Aladdin, HPLC, ≥99.9%) were used without any further purification. 1-Chlorobutane (AR), acetonitrile (HPLC), hydrochloric acid (AR), ethanol (AR) were purchased from Jiangtian Co. Ltd. and used as received.
Preparation of ionic liquids
The chlorostannate(II) ionic liquids were synthesized according to the published literature,29,30 and these ILs are shown in Scheme 1.
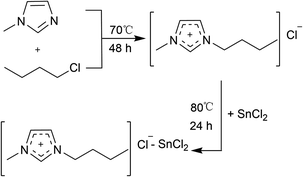 |
| | Scheme 1 Synthesis of the [Bmim]Cl–SnCl2 ILs. | |
Preparation of 1-butyl-3-methylimidazolium chloride [Bmim]Cl. The 0.24 mol (22.2 g) of 1-chlorobutane was added dropwise to a 100 ml four-necked round-bottom flask containing the 0.2 mol (16.4 g) of 1-methylimidazole. The reaction continued for 48 h at 70 °C. The excess 1-chlorobutane was removed by using a rotary evaporator under reduced pressure. [Bmim]Cl: 1H NMR (500 MHz, D2O) δ 8.55 (s, 1H), 7.31 (s, 1H), 7.26 (s, 1H), 4.03 (t, J = 7.1 Hz, 2H), 3.72 (s, 3H), 1.72–1.63 (m, 2H), 1.20–1.09 (m, 2H), 0.75 (t, J = 7.4 Hz, 3H); 13C NMR (126 MHz, D2O) δ = 136.03, 123.69, 122.45, 49.51, 36.02, 31.52, 19.01, 12.98.
Synthesis of [Bmim]Cl–SnCl2 systems. The precursor [Bmim]Cl (17.46 g, 0.1 mol) was charged into a 100 ml round-bottom flask equipped with an argon inlet and a thermometer. In order to synthesize the melts of different compositions (0.33 ≤ xSnCl2 ≤ 0.67), an appropriate amount of anhydrous SnCl2 powder was added batchwise at 80 °C under argon atmosphere. After the addition was completed, the mixture was stirred vigorously overnight to ensure complete reaction.In this work, the molar fraction (χ) of SnCl2 in the synthesized ILs is defined as
| |
 | (1) |
where
n1 is the number of moles of SnCl
2 and
n2 is the number of moles of [Bmim]Cl.
Analytical methods of ILs
[Bmim]Cl–SnCl2 systems were identified by 1H NMR spectrometer. NMR spectra were obtained on a Varian INOVA-500 spectrometer with DMSO as a solvent and TMS as an internal standard. The thermal stability of the [Bmim]Cl and [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) were determined by thermogravimetric analysis (TGA) at a heating rate of 10 K min−1 under nitrogen atmosphere from 25 to 500 °C. Raman spectra of the needle-shaped precipitate in [Bmim]Cl–SnCl2 (xSnCl2 = 0.67) were recorded using a RENISHAW InVia Reflex Raman spectrometer, with a 785 nm focused laser beam.
Evaluation of acidity of ILs
Yang and Kou31 have determined the acidity of ILs by means of an IR spectroscopic probe, wherein pyridine was used as a probe molecule for determination of the Lewis and Brønsted acidity of ILs and ethanenitrile, a weaker Lewis base than pyridine, was also used to measure the Lewis acidity of ILs. Acidity evaluation was carried out according to the following procedure: the samples were prepared by respectively adding pyridine and ethanenitrile to ILs in a given volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, and then these samples were analyzed by IR spectroscopy. The spectra were also recorded on the Bruker TENSON 27 FT-IR spectrometer. All spectra were acquired at a 4 cm−1 resolution with a total of 64 scans per spectrum.
:3, and then these samples were analyzed by IR spectroscopy. The spectra were also recorded on the Bruker TENSON 27 FT-IR spectrometer. All spectra were acquired at a 4 cm−1 resolution with a total of 64 scans per spectrum.
Reaction procedure and analysis
Synthesis of prepolymer. 100 g of L-lactic acid (90 wt% aqueous solution) was charged into a 250 ml four-necked flask equipped with a mechanical stirrer, a nitrogen gas inlet, a Vigreux column and a distillation head. The flask was heated at 100 °C for 1 h under nitrogen atmosphere; 130 °C for 1 h at a reduced pressure of 100 torr and finally at 150 °C for 1 h at a reduced pressure of 50 torr. The stirring speed was maintained at 120 rpm throughout the melt polymerization. Hereafter, the flask was cooled to room temperature, a viscous liquid of O-PLLA was obtained with 92.2% yield under nitrogen atmosphere. Nuclear magnetic resonance spectroscopy (NMR, 500 MHz, Varian) was used to measure the number average molecular weight. The samples were dissolved in solvent of chloroform (CDCl3). The average degree of polymerization (DP) of O-PLLA was 12 (see Fig. S2 in ESI†).
Synthesis of L-lactide. A 250 ml four-necked flask was equipped with a mechanical stirrer, a thermometer, a distillation condenser and a coiled receiver trap. The O-PLLA was charged into the flask and mixed with a predetermined amount of the IL catalyst. The flask was heated up to a required temperature at a predetermined pressure, in order for the crude lactide to be expelled by distillation and collected in the receiver container. The condenser was heated by circulating hot water (about 80 °C) to prevent crystallization of the distillate from sticking, while the receiver container was placed in an ice bath. The depolymerization was carried out for a period of time. The distillate (called crude lactide) was analyzed by using NMR spectroscopy (NMR, 500 MHz, Varian).
Purification of L-lactide. The crude lactide was washed with water at 0 °C for three times; then dried in vacuo for 48 h at 25 °C. The specific rotation of the dried product was immediately examined with an automatic polarimeter using a 0.01 g ml−1 toluene solution at 25 °C and a wave length of 589 nm. The optical purity of purified lactide was calculated using the equation| |
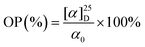 | (2) |
where [α]25D is the value of optical rotation measured with an automatic polarimeter and α0 is the specific optical rotation of 100% crystallized L-lactide.32
Recycling of PLLA residue for L-lactide resynthesis
The PLLA residue (PLLAr) was formed after lactide synthesis by the prepolymer route. A mixture of L-lactic acid (71.50 g, 0.71 mol, 90 wt% aqueous solution) and PLLAr (24.45 g, 0.34 mol) was heated at 120 °C under nitrogen atmosphere. The hydrolytic degradation was conducted for 24 h under continuous stirring to obtain monomeric lactic acid (DP < 2, calculated by comparing the integral areas of 1H NMR signals). Then, the L-lactide was resynthesized from the prepolymerization reaction step, according to Scheme 2.
 |
| | Scheme 2 Recycle process for L-lactide resynthesis. | |
Results and discussion
Characterizations of the ionic liquids
Synthesis of chlorostannate(II) ionic liquids. These chlorostannate(II) ionic liquids was prepared by addition of solid tin(II) chloride to solid [Bmim]Cl, which yielded colorless room-temperature ionic liquids for all xSnCl2 ≤ 0.63 compositions. When the sample for xSnCl2 = 0.67 was left to cool, a large amount of needle-shaped crystals grew within a few hours. The precipitate was obtained by filtration, and then dried at 100 °C in vacuum for 24 h. Raman spectra of the needle-shaped precipitate featured bands at 139(s), 166(s), and 192(s) cm−1, which corresponds to the Raman spectra of neat tin(II) chloride, with bands at 138(s), 163(s), and 193(s) cm−1, as shown in Fig. S1 (ESI†). It is noteworthy that, for xSnCl2 = 0.67, the phase behavior was thermally reversible; i.e., heating the xSnCl2 = 0.67 composition resulted in complete liquefaction, and subsequent cooling again resulted in the separation of needle-shaped crystals.
1H NMR analyses of the ionic liquids. Imidazolium-based chlorometallate ionic liquids could be studied using standard 1H NMR spectroscopy, particularly via the chemical shifts for C-2 proton on the imidazolium ring, which reveals the interactions of imidazolium cations with anions.33,34 1H NMR spectra were measured for [Bmim]Cl–SnCl2 systems at ambient temperature (see Fig. 1). It is worth pointing out that for xSnCl2 = 0.67 sample, the precipitate has been removed by filtration prior to the measurements.
 |
| | Fig. 1 Analyses of 1H NMR of ILs: (a) 1H NMR spectra of [Bmim]Cl and [Bmim]Cl–SnCl2 systems at xSnCl2 = 0.33, 0.50, 0.60 and 0.63, respectively; (b) changes in chemical shift of the acidic proton in the C-2 position of the imidazolium ring, as a function of composition, xSnCl2. | |
As demonstrated in Fig. 1a, the δC2–H signals are strongly shifted far upfield when compared to the chemical shifts of other protons. This demonstrates that the C-2 proton is the most sensitive towards variations of hydrogen -bond-accepting ability of the anions. The δC2–H for neat [Bmim]Cl is very high (10.27 ppm), which indicates that there is much more hydrogen bonding to the chloride anion than complex anions. Moving towards higher xSnCl2 values (xSnCl2 < 0.50), the δC2–H signals are shifted to higher fields, but the chemical shifts for xSnCl2 = 0.60–0.67 appear to be plateaued at around 9.1 ppm, as shown in Fig. 1b. Upon addition of tin(II) chloride, changes of δC2–H signals disclose that the strength of the cation–anion interactions decreases. This could be interpreted in terms of the formation of complex anion clusters. To the best of our knowledge, the existence of two chlorostannate(II) anions: [SnCl3]−and [Sn2Cl5]−, has been demonstrated.29 For xSnCl2 = 0.50, the δC2–H signal is shifted to 9.37 ppm, since that electron-rich anions ([SnCl3]−) are present, resulting in shielding of the H nucleus. The progressive addition of tin(II) chloride led to a gradual decrease of the chemical shift for xSnCl2 = 0.50–0.60. Above this concentration (xSnCl2 = 0.60), it is possible that the [Sn2Cl5]− dimer is formed, which leads to further weakening of cation–anion interactions, because of charge distribution of a single negative charge over a greater number of electronegative centres. But there are not enough dimers present for H-bonding to each imidazolium cation in [Bmim]Cl–SnCl2 systems, due to the formation of tin(II) chloride precipitate for xSnCl2 > 0.63 samples. It appears that the composition of [Bmim]Cl–SnCl2 systems for xSnCl2 ≥ 0.63 remained unchanged. In a word, for xSnCl2 = 0.50, only [SnCl3]− anion is present in [Bmim]Cl–SnCl2 systems; for xSnCl2 > 0.50, there is two kinds of chlorostannate(II) anions: [SnCl3]−and [Sn2Cl5]−. It is worth noting that [Bmim]Cl–SnCl2 systems based solely on [Sn2Cl5]− clusters as anions are unstable and inexistent at room temperature.
TG analysis of [Bmim]Cl and [Bmim]Cl–SnCl2 (xSnCl2 = 0.63). The [Bmim]Cl and [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) ionic liquids were tested for their thermal stability by TGA in the temperature range between 25 and 500 °C. By differential computing the TG curves, the derivative thermogravimetric (DTG) curves were obtained. As can be seen from Fig. 2, extrapolated starting temperature (Tei) used as the decomposition temperature, was estimated. For the both ionic liquids, the decomposition temperature increases from 268 °C ([Bmim]Cl) to 390 °C (xSnCl2 = 0.63). The TGA results indicate that the thermal stability of [Bmim]Cl ionic liquid has been enhanced by coordinating chlorostannate(II) anions.
 |
| | Fig. 2 TG and DTG curves of [Bmim]Cl and [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) at a heating rate of the TG apparatus of 10 K min−1 in the temperature range 25–500 °C. | |
Evaluation of acidity of the ionic liquids
Evaluation of acidity by IR acetonitrile spectroscopic probe. Acetonitrile has been used as a FT-IR spectroscopy probe to characterize the Lewis acidic strength.31 The nitrile group is reacted with the Lewis acid to produce the CN-Lewis complex, which shows a new band at 2200–2400 cm−1. With the increase of the Lewis acidic strength, this band moves to higher wavenumbers.31,35 Hereby, acetonitrile was added to the synthesized ILs followed by FT-IR scanning to determinate the Lewis acidic strength of ILs.As shown in Fig. 3, the bands of acetonitrile/[Bmim]Cl and acetonitrile/[Bmim]Cl–SnCl2 (xSnCl2 = 0.33) were nearly the same as that of acetonitrile. They showed two characteristic bands at approximately 2295 and 2250 cm−1, originating from its CN stretching vibrations, indicative of the lack of Lewis acidity of [Bmim]Cl and [Bmim]Cl–SnCl2 (xSnCl2 = 0.33). However, in the spectra of acetonitrile/[Bmim]Cl–SnCl2 (xSnCl2 = 0.50), acetonitrile/[Bmim]Cl–SnCl2 (xSnCl2 = 0.60) and acetonitrile/[Bmim]Cl–SnCl2 (xSnCl2 = 0.63), new absorption peaks at around 2330 cm−1 appeared. The new absorption peak was the characteristic absorption peak of the CN-Lewis complex, indicating that [Bmim]Cl–SnCl2 (xSnCl2 = 0.50), [Bmim]Cl–SnCl2 (xSnCl2 = 0.60) and [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) had Lewis acidity. Moreover, when xSnCl2 > 0.50, a monotonic blue shift of new bands was observed with increasing value of xSnCl2, corresponding to an increase in Lewis acidic strength.
 |
| | Fig. 3 FT-IR spectra of ILs using acetonitrile as probe. (a) pure acetonitrile; (b) acetonitrile + [Bmim]Cl; (c) acetonitrile + [Bmim]Cl–SnCl2 (xSnCl2 = 0.33); (d) acetonitrile + [Bmim]Cl–SnCl2 (xSnCl2 = 0.50); (e) acetonitrile + [Bmim]Cl–SnCl2 (xSnCl2 = 0.60); (f) acetonitrile + [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) (acetonitrile/ionic liquid = 1:3 by volume in b–f). | |
Evaluation of acidity by IR pyridine spectroscopic probe. As a FT-IR spectroscopy probe, pyridine can react with Brønsted and Lewis acids. A band near 1540 cm−1 is an indication of the formation of the cation [PyH]+ resulting from the presence of Brønsted acid sites, while a band near 1450 cm−1 is an indication of pyridine coordinated to Lewis acid sites.31,36 By observing these two peaks, the acidic type of the samples can be deduced. Therefore, this method was also used to characterize [Bmim]Cl–SnCl2 (xSnCl2 = 0.50) and [Bmim]Cl–SnCl2 (xSnCl2 = 0.63). As shown in Fig. 4, when pyridine was mixed with [Bmim]Cl–SnCl2 (xSnCl2 = 0.50), no band at 1540 cm−1 was observed, suggesting that there was no Brønsted acidity. Two bands of 1540 and 1446 cm−1 appeared in the FT-IR spectra of pyridine/[Bmim]Cl–SnCl2 (xSnCl2 = 0.63), indicating that [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) contained Brønsted acidity and Lewis acidity.
 |
| | Fig. 4 FT-IR spectra of ILs using pyridine as probe. (a) Pure pyridine; (b) pyridine + [Bmim]Cl–SnCl2 (xSnCl2 = 0.50); (c) pyridine + [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) (pyridine/ionic liquid = 1:3 by volume in b–c). | |
L-Lactide synthesis
The sample of O-PLLA, with the average degree of polymerization of 12 (see Fig. S2 in ESI†), was synthesized. When the depolymerization reaction was carried out using O-PLLA (DP = 12), the effect of different catalyst systems on the yield of crude lactide was studied (see Fig. 5). The distillate solid is defined as crude lactide, containing D-lactide, L-lactide, meso-lactide, and other impurities such as lactic acid, lactoyl lactic acid and other acyclic. The yield is expressed by eqn (3):| |
 | (3) |
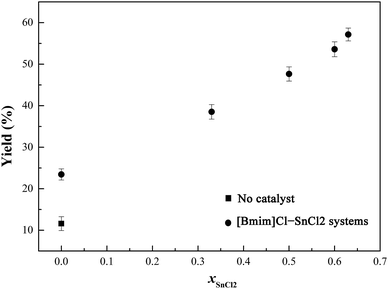 |
| | Fig. 5 Effect of [Bmim]Cl–SnCl2 catalyst systems on the yield of the crude lactide. Reaction conditions: 200 °C, 10 torr, 2 h, O-PLLA (DP = 12), 300 rpm, catalyst 0.1 mol% relative to O-PLLA. | |
As can be seen from Fig. 5, using the catalyst multiplied the yield of crude lactide, and as to the [Bmim]Cl–SnCl2 catalyst systems, the crude lactide was obtained in moderate to good yield (39%–57%). Meanwhile, the yield increased significantly as the molar fraction of SnCl2 increased from 0 to 0.63. This was mainly because with an increased amount of SnCl2, the acidity of the ionic liquids became stronger. These catalysts were uniformly dispersed in the mixture and sufficiently contacted with the reactants, because they were miscible with O-PLLA at a catalytic amount throughout the depolymerization process. Synthesis of L-lactide is also summarized in Table 1, using SnCl2, [Bmim]Cl, or [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) as a catalyst, respectively. D,L-lactide means a minute amount of D-lactide in addition to L-lactide. The fractional amount of D,L-lactide in crude lactide could be calculated by comparing the integral areas of 1H NMR signals (see Fig. S3 in ESI†). The results (Table 1) indicated that D,L-lactide fraction was significantly increased by using IL catalyst. It meaned that the presence of ILs reduced or eliminated the negative impact of side reactions. It is known that, meso-lactide, lactic acid and acyclic oligomers are more soluble in water than L-lactide. Thus, water-washing method was conducted to obtain the pure lactide.
Table 1 Synthesis of L-lactide by depolymerization of O-PLLAa
| No. |
Catalyst |
Lactide synthesis |
Residue |
| D,L-lactideb |
OPc |
DPd |
Iso.e |
| 200 °C, 10 torr, 2 h, O-PLLA (DP = 12), 300 rpm, catalyst 0.1 mol% relative to O-PLLA. Calculated by 1H NMR. (see Fig. S3 in ESI). Measured with an automatic polarimeter. Measured with 1H NMR. (see Fig. S5 in ESI). Isotacticity measured with 13C NMR. |
| 1 |
SnCl2 |
77.5% |
90.0% |
56 |
78.5% |
| 2 |
[Bmim]Cl |
91.2% |
98.4% |
52 |
95.4% |
| 3 |
[Bmim]Cl–SnCl2(xSnCl2 = 0.63) |
97.9% |
99.9% |
49 |
99.1% |
When using [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) as a catalyst, the optical purity of purified lactide was 99.9%, which was much higher than using SnCl2. As for the isotacticity of residues, the same situation occurred. This was probably because the formation of the complex clusters (e.g., [SnCl3]−, [Sn2Cl5]−) enhanced the acidity of the anion, then decreased the degree of the racemization that usually occurred via the α-proton abstraction.17
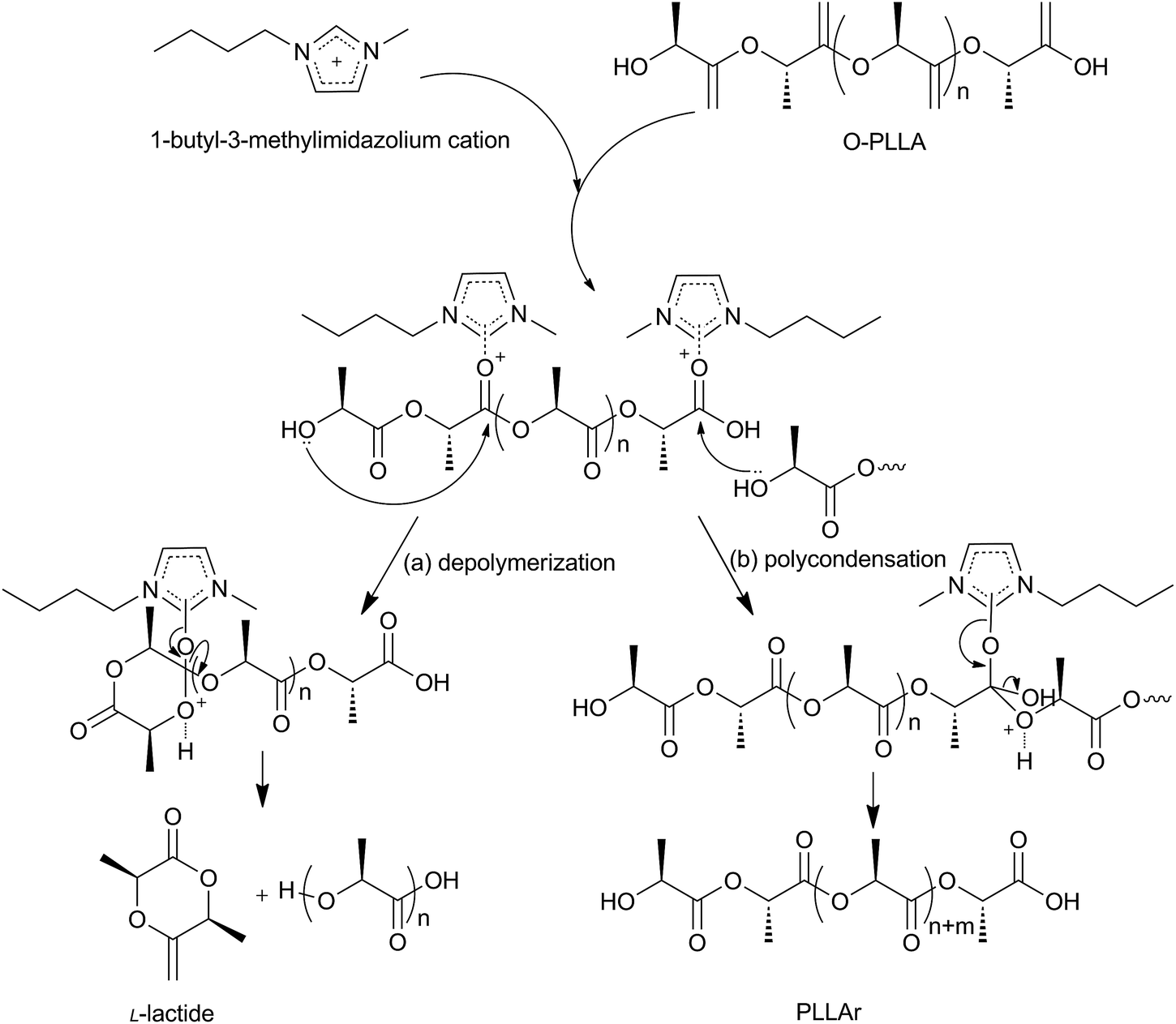
It is clear that the synthetic system of L-lactide involves two reaction equilibria: depolymerization equilibrium and polycondensation equilibrium of partial O-PLLA molecules. The L-lactide is produced by the depolymerization of O-PLLA, which is actually an intra-molecular transesterification reaction.37–39 Based on the structure of ILs (as shown in Scheme 1), we proposed a possible mechanism, as illustrated in Scheme 3. The imidazolium cation forms a tight association with the carbonyl oxygen so as to enhance the electrophilicity of the carbonyl-carbon. Then, the attack of a hydroxyl terminal of O-PLLA molecule upon the carbonyl-carbon followed by the intra-molecular transesterification leads to the formation of six-membered cyclic ester. It should be mentioned that, the counter anion of the imidazolium cation is the terminal carboxylate in the O-PLLA chain and the steric hindrance of the terminal carboxylate group is very high. By this means, the chance of the possible racemization via α-proton abstraction17,19 is greatly reduced. The isotacticity of the residual polymers has proved this point (for [Bmim]Cl–SnCl2 (xSnCl2 = 0.63), Iso. = 99.1%; for SnCl2, Iso. = 78.5%), as listed in Table 1.
 |
| | Scheme 3 Possible mechanism of depolymerization – polycondensation behavior of O-PLLA catalyzed by 1-butyl-3-methylimidazolium ionic liquids. | |
According to the above mechanism, both the strength of the cation–anion interaction and the Lewis acidic strength of ILs play important roles in the catalytic activity. The strong cation–anion interaction hinders the imidazolium from getting close to the O-PLLA, thus reducing the catalytic activity. ILs incorporating stannum(II) weaken the cation–anion interaction and enhance the Lewis acidity of the imidazolium, resulting in higher catalytic activity. These considerations are in agreement with our experimental results.
Effects of various reaction parameters
The effects of various reaction parameters, such as temperature, reaction time, pressure and mole ratio of catalyst/O-PPLA, were investigated in detail.
Fig. 6 displays the effect of the reaction temperature on the yield of crude lactide in the temperature range of 180–240 °C. As the reaction temperature increased, the yield went up gradually due to the fact that the depolymerization reaction rate increased with increasing temperature. As far as we know, the side reactions such as racemization, elimination or formation of the ether derivatives, are accelerated by increased temperature.17,20 In order to ensure the chemical and optical purity of L-lactide, the results indicated that 200 °C was an appropriate reaction temperature for [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) ionic liquid.
 |
| | Fig. 6 Variation of yield% as a function of reaction temperature. Reaction conditions: 10 torr, 2 h, O-PLLA (DP = 12), 300 rpm, [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) 0.1 mol% relative to O-PLLA. | |
The effect of reaction time on crude lactide yield was studied by varying the reaction time from 0 to 4 h. As illustrated in Fig. 7, the yield obviously increased with increasing of reaction time from 0 to 2 h. However, a slight increase in the yield was observed when the reaction time was increased from 2–4 h. Hence, we chose 2 h as the suitable reaction time in the subsequent experiments.
 |
| | Fig. 7 Variation of yield% as a function of reaction time. Reaction conditions: 200 °C, 10 torr, O-PLLA (DP = 12), 300 rpm, [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) 0.1 mol% relative to O-PLLA. | |
Fig. 8 shows the variation of crude lactide yield as a function of pressure. The yield increased from 26 to 57% as the pressure was decreased from 100 to 10 torr. After the lactide was formed, it was distilled immediately at the lower pressure, thus increasing the conversion.
 |
| | Fig. 8 Variation of yield% as a function of pressure. Reaction conditions: 200 °C, 2 h, O-PLLA (DP = 12), 300 rpm, [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) 0.1 mol% relative to O-PLLA. | |
Fig. 9 shows the influence of the catalyst concentration on the yield of crude lactide. The yield increased rapidly from 48 to 60% as the mole ratio of [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) to O-PLLA increased from 0.025 to 0.1. The higher yield obtained at a higher catalyst concentration may have been due to the higher reaction kinetics. However, the further increase in the mole ratio would not cause an obvious increase in the yield of crude lactide. Therefore, the optimal mole ratio was 0.1.
 |
| | Fig. 9 Variation of yield% as a function of mole ratio of [Bmim]Cl–SnCl2 (xSnCl2 = 0.63)/O-PPLA. Reaction conditions: 200 °C, 10 torr, 2 h, O-PLLA (DP = 12), 300 rpm. | |
Recycle of PLLAr for L-lactide resynthesis
During the process of synthesizing L-lactide by the prepolymer route, the high-Mw oligomer residue is inevitably formed. The reasonable recycle of these residual polymers is important in both academia and industry as viewed from the cost reduction and environment protection. When using [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) as a catalyst, the residual polymer of DP = 49 was formed and it possessed high isotacticity of 99.1% that was much higher than using the conventional SnCl2 catalyst (Table 1). Obviously, this makes it possible to realize the reutilization of these residues for the resynthesis of L-lactide (Table 2).
Table 2 Recycle of PLLAr for L-lactide resynthesisa
| Rec.b |
Hydrolysis step |
Prepolymerization step |
Depolymerization step |
| L-LA replenished |
O-PLLA (DP = 12) |
L-Lactide |
Residue |
| wt (g) |
molc |
wt (g) |
molc |
Oligomer yield |
wt (g) |
molc |
Yieldd |
Yielde |
wt (g) |
DPf |
Iso.g |
| 200 °C, 10 torr, 2 h, 300 rpm, [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) 0.1 mol% relative to O-PLLA. Recycle time. Based on lactic acid unit. Base on O-PLLA. Base on L-LA replenished. Measured with 1H NMR. Isotacticity measured with 13C NMR. |
| 0 |
100.00 |
1.00 |
67.76 |
0.92 |
92.2% |
38.76 |
0.54 |
57.2% |
|
24.45 |
49 |
99.1% |
| 1 |
71.50 |
0.71 |
72.87 |
0.99 |
94.2% |
42.78 |
0.59 |
58.7% |
83.1% |
26.80 |
54 |
98.8% |
| 2 |
78.55 |
0.78 |
79.12 |
1.08 |
93.1% |
44.90 |
0.62 |
56.7% |
79.4% |
26.15 |
46 |
98.6% |
| 3 |
76.32 |
0.76 |
76.57 |
1.04 |
92.6% |
44.00 |
0.61 |
57.5% |
80.1% |
25.48 |
52 |
98.5% |
The residual polymer was hydrolyzed with aqueous lactic acid (90 wt%) to regenerate monomeric lactic acid. The amount of water in the added lactic acid is 1.2 mole equivalent to hydrolyze PLLAr. The hydrolysate was successively subjected to oligomerization and depolymerization to resynthesize L-lactide. As shown in Table 2, the reiterative L-lactide synthesis proceeded as efficiently as the first run to give stable yields of 56.7%–58.7% (Based on O-PLLA). The loss in the oligomer yield was due to vaporization of the unreacted L-lactic acid and oligomer with low molecular weight less than 500 g mol−1. Notably, the [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) catalyst was entirely contained in the high-Mw oligomer residues because of its high thermal stability (Td = 390 °C). Thus, the reiterative depolymerization was conducted without further addition of the catalyst. Under the same reaction conditions, [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) catalyst showed no notable loss of activity up to three cycles. It was worth noting that, in this process, the yield calculated on the basis of the replenished L-lactic acid after the second cycle reached up to 80–83%, which indicated the practicability of the present recycle process. In addition, the isotacticity of PLLAr was maintained at a high level during the recycle process. Hence, [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) is thought to be an efficient and recyclable catalyst for L-lactide production by the prepolymer route.
Conclusions
In this study, it is the first time that chlorostannate(II) ionic liquids have been used as catalyst for depolymerization of O-PLLA into L-lactide with high chemical and optical purity. The catalytic activity increases with the more ratio of SnCl2 increase in the ILs. Meanwhile, the experimental results showed that the optimum reaction conditions (200 °C, 10 torr, 2 h, 300 rpm, [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) 0.1 mol% relative to O-PLLA) were obtained, resulting in that L-lactide is white crystalline powder with little impurity (<0.1%). Because the use of these ionic liquids minimizes the contamination of a reactive group with by-products, the use of recrystallization for purification of L-lactide is unnecessary, which would be helpful for simplifying the production process of enantiomerically pure lactide. Further, owing to a high boiling point of [Bmim]Cl–SnCl2 (xSnCl2 = 0.63), this catalyst can be easily recycled. The residue polymers of high isotacticity were utilized to resynthesize L-lactide for 3 circles without loss of the yield of lactide. Remarkably, the lactide yield reached to about 80% (based on L-LA replenished). The results show the feasibility of using [Bmim]Cl–SnCl2 (xSnCl2 = 0.63) as a catalyst for the resynthesis of high-quality L-lactide. This procedure would contribute to reduce the cost of the synthetic process of high-quality L-lactide, owing to the recycle of PLLAr.
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (no. 21336007).
Notes and references
- K. M. Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493 CrossRef PubMed.
- A. K. Mohanty, M. Misra and G. Hinrichsen, Macromol. Mater. Eng., 2000, 276, 1 CrossRef.
- W. Groot, in Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, And Applications, ed. R. Auras, L.-T. Lim, S. E. M. SelkeandH. Tsuji,John Wiley & Sons, Inc., 2010, ch. 1, pp. 3–16 Search PubMed.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820 CrossRef CAS PubMed.
- S. I. Moon, C. W. Lee, M. Miyamoto and Y. Kimura, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1673 CrossRef CAS.
- H. Li, C. H. Wang, F. Bai, J. Yue and H. G. Woo, Organometallics, 2004, 23, 1411 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS PubMed.
- Y. Yoshiaki and A. Tomohiro, US Pat., 5502215, 1996.
- B. B. Idage and S. Swaminathan, US Pat., 20130060051A1, 2012.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63 CrossRef CAS.
- S. I. Moon, C. W. Lee, I. Taniguchi, M. Miyamoto and Y. Kimura, Polymer, 2001, 42, 5059 CrossRef CAS.
- K. Hirao and H. Ohara, Polym. Rev., 2011, 51, 1 CrossRef CAS PubMed.
- Y. Liu, R. Wei, J. Wei and X. Liu, Progr. Chem., 2008, 20, 1588 CAS.
- M. Noda and H. Okuyama, Chem. Pharm. Bull., 1999, 47, 467 CrossRef CAS.
- H. Tsuji, I. Fukui, H. Daimon and K. Fujie, Polym. Degrad. Stab., 2003, 81, 501 CrossRef CAS.
- M. Noda, Prep. Biochem. Biotechnol., 1999, 29, 333 CrossRef CAS PubMed.
- D. K. Yoo, D. Kim and D. S. Lee, Macromol. Res., 2006, 14, 510 CrossRef CAS.
- M. Ehsani, K. Khodabakhshi and M. Asgari, e-Polym., 2014, 14, 353 CAS.
- W. Huang, Y. Qi, N. Cheng, X. Zong, T. Zhang, W. Jiang, H. Li and Q. Zhang, Polym. Degrad. Stab., 2014, 101, 18 CrossRef CAS PubMed.
- Y. Ishijima, Y. Maruta and A. Abiko, Chem. Lett., 2012, 41, 1456 CrossRef CAS.
- J. Estager, J. D. Holbrey and M. Swadzba-Kwasny, Chem. Soc. Rev., 2014, 43, 847 RSC.
- A. W. Taylor, S. Men, C. J. Clarke and P. Licence, RSC Adv., 2013, 3, 9436 RSC.
- H. Iwahashi, T. Oka and A. Abiko, Chem. Lett., 2008, 37, 708 CrossRef CAS.
- W. Wang, L. Wu, Y. Huang and B. G. Li, Polym. Int., 2008, 57, 872 CrossRef CAS PubMed.
- S. Zhang, H. Lefebvre, M. Tessier and A. Fradet, Green Chem., 2011, 13, 2786 RSC.
- S. Mallakpour and M. Dinari, Iran. Polym. J., 2011, 20, 259 CAS.
- S. H. Kim, C. H. Hong, D. S. Han and J. Y. Seo, US Pat., 2014187798A1, 2014.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- M. Currie, J. Estager, P. Licence, S. Men, P. Nockemann, K. R. Seddon, M. Swadzba-Kwasny and C. Terrade, Inorg. Chem., 2013, 52, 1710 CrossRef CAS PubMed.
- P. Illner, A. Zahl, R. Puchta, N. van Eikema Hommes, P. Wasserscheid and R. van Eldik, J. Organomet. Chem., 2005, 690, 3567 CrossRef CAS PubMed.
- Y. L. Yang and Y. Kou, Chem. Commun., 2004, 226 RSC.
- P. L. Salzberg, US Pat., 2758987, 1956.
- A. Elaiwi, P. B. Hitchcock, K. R. Seddon, N. Srinivasan, Y.-M. Tan, T. Welton and J. A. Zora, J. Chem. Soc., Dalton Trans., 1995, 3467 RSC.
- J. A. McCune, P. He, M. Petkovic, F. Coleman, J. Estager, J. D. Holbrey, K. R. Seddon and M. Swadzba-Kwasny, Phys. Chem. Chem. Phys., 2014, 16, 23233 RSC.
- Z. K. Zhao, B. Yuan, W. H. Qiao, Z. S. Li, G. R. Wang and L. Cheng, J. Mol. Catal. A: Chem., 2005, 235, 74 CrossRef CAS PubMed.
- X. H. Wang, G. H. Tao, Z. Y. Zhang and Y. Kou, Chin. Chem. Lett., 2005, 16, 1563 CAS.
- T. Motoyama, T. Tsukegi, Y. Shirai, H. Nishida and T. Endo, Polym. Degrad. Stab., 2007, 92, 1350 CrossRef CAS PubMed.
- C. L. Sungyeap Hong, Mod. Chem. Appl., 2014, 02, 144 Search PubMed.
- S. A. Mulyashov, F. S. Sirovski and S. G. Beksaev, Chem. Eng. J., 2011, 176, 225 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Raman spectra (Fig. S1), molecular weight determination of O-PLLA (Fig. S2), calculation the fractional amount of D,L-lactide (Fig. S3), isotacticity measured with 13C NMR (Fig. S4), synthesis of PLLA by using ILs catalysts (Table S1), 1H NMR spectra of PLLA residues (Fig. S5). See DOI: 10.1039/c5ra05073a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.