
The distinct role of the flexible polymer matrix in catalytic conversions over immobilised nanoparticles†‡
Stefano Martinuzziab,
Daniela Cozzulab,
Paulo Centomoa,
Marco Zecca*a and
Thomas E. Müller*b
aUniversita' degli Studi di Padova, Dipartimento di Scienze Chimiche, via F. Marzolo 1, Padova, Italy. E-mail: marco.zecca@unipd.it; Tel: +39 049 8275737
bCAT Catalytic Center, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: Thomas.Mueller@catalyticcenter.rwth-aachen.de; Fax: +49 241 8022593; Tel: +49 241 8028594
First published on 15th June 2015
Abstract
Polymer resins with immobilized metal nanoparticles represent highly promising materials for attaining intelligent and ecologically friendly catalysts. Control of the dynamic swelling behaviour of the polymer resins enables the on/off-switching of the activity of the encapsulated metal nanoparticles, thereby allowing conversion and selectivity to be controlled at a specific time of the chemical reaction. This paper presents a study on the distinct role of the polymer support. Here, the hydrogenation of nitrobenzene was chosen as a model reaction, and nanoparticles of different metals were taken into account. The course of the reaction revealed the essential role of the polymer resins in controlling the diffusion, in particular of the reagents, to and from the catalytically active sites on the surface of the nanoparticles thereby influencing the catalytic activity of the metal and opening up a new approach to catalytic engineering.
Introduction
Recently, the use of metal nanoparticles1 on polymer supports has attracted growing interest in reactions such as the anthraquinone process,2 the direct synthesis of H2O2,3 the hydrogenation of nitrobenzene,4,5 cyclohexene and α,β-unsaturated aldehydes,6 styrenes,7 the selective reduction of benzene to cyclohexene,8 dehalogenation of chlorobenzene,9,10 as well as in the conversion of glucose to sorbose.11 Yet these applications12–15 are few compared to the large number of applications of oxidic supports widely used to immobilize metal nanoparticles.1,16–18 The increasing attention paid to polymer matrices as a support for nanoparticles is related to the high flexibility of the polymer resins, but also to the facile preparation and the ready property control which are superior features compared to many of the classic supports. Interestingly, polymer supports provide new opportunities in controlling catalytic reactions.Aiming at developing novel smart catalysts,19–21 this study investigates the use of polymeric supports22–26 to stabilise metal nanoparticles.27–29,35 The focus was learning how to switch the activity of catalysts on/off, expanding the scope of polymer supports and more generally introducing smart heterogeneous catalysts in the liquid-phase synthesis of fine chemicals. The objective of this study is to promote understanding of the effect of the reaction medium on the swelling behaviour of polymer supports, thereby changing the dimensions of the porous polymer network and, consequently, the properties of the resin.
Dry gel-type resins are glassy materials with no intrinsic porosity. The pore network develops only upon swelling of the polymer by a liquid host, the nature of which strongly affects the pore size. The degree of swelling is relevant to both catalyst preparation and during the use of polymer supported catalysts in catalytic reactions. The sulphonated gel-type ion-exchange resin K1221 was chosen as the polymer support (Fig. 1).30 The resin is characterised by high acidity (due to the presence of –SO3H groups with pKa = −2.8 in water) and good swelling properties in aqueous media. As it is readily recovered, the resin K1221 has found wide applications in water purification. It is also employed in industrial applications, when a strong acid is needed, but no free acid in solution is allowed, as for the hydrolysis of ethers and esters,31 condensation reactions involving small polar molecules32 and bisphenol-A production.33 A particular advantage of the K1221 polymer beads is the interconnected network that allows to control the size of the metal nanoparticles formed inside the network.34,35 It is well known that the metal particles tend to grow in size,1,29,36,37 until the available space between the polymer chains has been filled (Scheme 1).
 | ||
| Fig. 1 Chemical structure of the K1221 ion exchange resin (left) and image of the parent K1221 beads (right). | ||
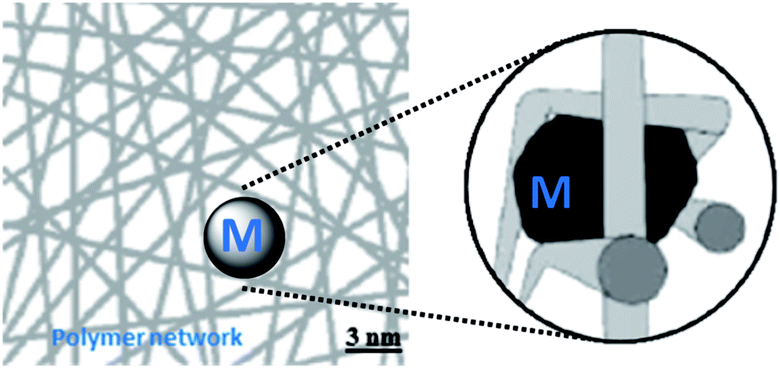 | ||
| Scheme 1 Restricted growth of metal nanoparticles embedded in a gel-type resin.35 | ||
In this study, different catalysts were synthesized by growing nanoparticles of metals such as Rh, Pt, Ru, and Ni into the water-swollen polymer resin. We chose to concentrate on nitrobenzene hydrogenation as model reaction, as this reaction is characterized by a very different polarity of the nitrobenzene substrate, the aniline intermediate and the cyclohexylamine product. Thus, we expected to obtain insight into how the solvent and the changing polarity of the reaction medium affect the accessibility of catalytic sites inside the polymer network.
Experimental
All chemicals were obtained from commercial suppliers and used as received if not stated otherwise. The K1221 resin (bead size 261(5) μm, bulk density 760 g l−1, ion exchange capacity 1.9 mmol g−1) was obtained from Lanxess; active carbon was obtained from Evonik.Catalyst preparation
The polymer-supported catalysts‡ were prepared by an impregnation procedure. The parent K1221 resin was washed with deionized water, the sulfonic acid groups neutralized with aqueous sodium hydroxide (1 mol l−1) and the beads washed with water until the pH-value of the effluent was neutral. Subsequently, the beads were washed with MeOH/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as well as with neat MeOH and dried for 24 h at 80 °C. The sodium exchanged beads (5.83 g) were suspended in water (30 ml) for 2 h. A solution of the metal salt was added (Table 1, Step 1); the mixture stirred at RT for 3 h and then heated to reflux for 30.5 h. After cooling to RT, the beads were washed with deionized water. The beads were re-suspended in water (5 ml), and a solution of NaBH4 (Table 1, Step 2) was added under vigorous stirring. The mixture was stirred for 22 h. Then the beads were filtered off, washed with ample deionized water and then dried in air at 70–85 °C for 6 d. The metal content was 5.3%w/w (Rh/K1221), 8.6%w/w (Pt/K1221), 5.7%w/w (Ru/K1221), 2.4%w/w (Ni/K1221).
:1 v/v) as well as with neat MeOH and dried for 24 h at 80 °C. The sodium exchanged beads (5.83 g) were suspended in water (30 ml) for 2 h. A solution of the metal salt was added (Table 1, Step 1); the mixture stirred at RT for 3 h and then heated to reflux for 30.5 h. After cooling to RT, the beads were washed with deionized water. The beads were re-suspended in water (5 ml), and a solution of NaBH4 (Table 1, Step 2) was added under vigorous stirring. The mixture was stirred for 22 h. Then the beads were filtered off, washed with ample deionized water and then dried in air at 70–85 °C for 6 d. The metal content was 5.3%w/w (Rh/K1221), 8.6%w/w (Pt/K1221), 5.7%w/w (Ru/K1221), 2.4%w/w (Ni/K1221).
| Metal | Step | Reagent | Quantity | Water | |
|---|---|---|---|---|---|
| [g] | [mmol] | [ml] | |||
| a x + y = 3, aqueous solution, 1.5%w/w of Ru.b Aqueous solution, 10%w/w of Rh, 5%w/w HNO3, 2.727 g solution used. | |||||
| Rh | 1 | [Rh(NO3)3(H2O)x]b | 2.727 | 2.65 | 2 |
| 2 | NaBH4 | 2.651 | 70.1 | 10 | |
| Pt | 1 | [Pt(NH3)4](NO3)2 | 0.609 | 1.57 | 15 |
| 2 | NaBH4 | 1.047 | 27.7 | 5 | |
| Ru | 1 | [Ru(NO)(NO3)x(OH)y]a | 18.71 | 2.78 | 7 |
| 2 | NaBH4 | 1.155 | 30.5 | 5 | |
| Ni | 1 | Ni(OAc)2·4H2O | 1.171 | 4.61 | 15 |
| 2 | NaBH4 | 2.083 | 55.1 | 10 | |
| Catalyst | Initial ratea [103 molNB molM−1 h−1] | Conv.b [%] | Selectivityb [%] | |||||
|---|---|---|---|---|---|---|---|---|
| NB | ANI | CHA | DCHA | ANI | CHA | DCHA | ||
| a 120 °C, 100 bar, 500 s m−1, THF/MeOH 9:1.b After 360 min.c Initiation period 1–5 min.d Change in regime (see text).e Parallel and consecutive product.f Consecutive product NB: nitrobenzene; ANI: aniline; CHA: cyclohexylamine; DCHA: dicyclohexylamine. |
||||||||
| Rh/K1221 | −1.6 | 1.4 | 0.1 | 0.005 | 82 | 81 | 18 | 0.9 |
| Rh/C | −4.1 | 1.9 | 1.9 | 0.4 | 100 | 42 | 47 | 11 |
| Pt/K1221 | −1.7 | 1.5 | 0.2 | 0.01 | 83 | 77 | 22 | 1.0 |
| Pt/C | −6.5c | 6.4 | 0f | 0.04 | 100 | 92 | 7 | 0.5 |
| Ru/K1221 | −1.2 | 1.0 | 0.2 | 0.01 | 72 | 76 | 22 | 1.7 |
| Ru/C | −1.3c,d | 1.2 | 0.1e | 0.7 | 100 | 20 | 66 | 14 |
| Ni/K1221 | −1.6 | 1.3 | 0.2 | 0.01 | 74 | 77 | 21 | 1.9 |
| Ni/C | −2.7c,d | 2.6 | 0.003 | 0.005 | 100 | 93 | 7 | 0.1 |
Carbon-supported benchmark catalysts were obtained from Sigma-Aldrich (5%w/w). Ni/K1221 was prepared by adding active carbon (4.753 g) to a solution of the metal salt (Table 1) in MeOH (30 ml). The mixture was stirred for 24 h. Then, the volatiles were removed in a partial vacuum, and the powder was maintained at 85 °C for another 2 h. The powder was then calcined in Argon at 300 °C (10 °C min−1 to 200 °C, isothermal for 60 min, 10 °C min−1 to 300 °C, isothermal for 60 min) and reduced in hydrogen (300 °C for 120 min, 1 °C min−1 to RT).
Nitrobenzene hydrogenation
Hydrogenation reactions‡ were performed in a 200 ml stainless steel autoclave equipped with gas entrainment stirrer (600 rpm). The autoclave was charged with catalyst beads, MeOH (12 ml) and dodecane (1.3 g), closed and flushed three times with Argon. After stirring the mixture for 30 min at RT, the autoclave was pressurized with hydrogen to 20 bar. By using a HPLC pump, THF (102 ml) was added. The mixture was then heated to the appropriate reaction temperature reported in the text. The pressure was adjusted with hydrogen to 100 bar and the reaction started by addition of nitrobenzene (2.5 g). The pressure was maintained thereafter at 100 bar by feeding hydrogen. In periodic intervals, samples of the liquid phase were taken and analysed by gas chromatography.Results and discussion
Catalyst preparation and characterization
Polymer-supported catalysts were prepared by impregnating the sodium exchanged K1221 beads in aqueous solution with a suitable Rh, Pt, Ru, and Ni precursor followed by reduction with NaBH4.‡ To characterise the porosity of the parent K1221 polymer matrix, a sample equilibrated in aqueous phase was investigated by Inverse Steric Exclusion Chromatography (ISEC).38,39 With this technique solutes of known molecular size are eluted through an HPLC column packed with the swollen resin. Assuming that the partition of the solutes between the mobile and the stationary phase is driven by steric factors only, the characteristic dimensions of the pore system in the stationary phase can be obtained by analysis of the elution volumes as a function of the molecular size of the eluates. Water was chosen for the analysis as an aqueous solution was employed for both, introduction of the metal precursors into the resin and during subsequent reduction of the precursor to the elemental metal with NaBH4. For this reason, the pore size and pore size distribution of water-swollen K1221 is expected to be relevant to the final size of the metal particles. To suppress enthalpic interactions in the partition process of the ISEC analysis, an electrolyte (Na2SO4, 0.2 M) was added to the mobile phase. The results of the ISEC analysis on the water-swollen K1221 are shown in Fig. 2. | ||
| Fig. 2 Pore size distribution of water-swollen K1221 (cylindrical pore model). | ||
The pores of water-swollen K1221 were modelled as cylindrical pores, which were found to fall in the range of small mesopores up to 8 nm in diameter, with a volume-average value of 3.4 nm. We therefore expected that the metal nanoparticles fall in the same size range.40,19 The size of the Rh, Pt, Ru, and Ni nanoparticles formed inside the K1221 network was explored by X-ray diffraction (Fig. 3). For Rh/K1221, three well resolved signals at 2θ values of 41°, 69° and 84° are assigned to the (111), (200) and (311) planes of the fcc crystal structure of rhodium. From the line broadening, an average size of the metal nanoparticles of 4.7 nm was calculated. The X-ray diffraction pattern of Pt/K1221 was very similar to Rh/K1221, showing three well resolved peaks at 2θ values of 39.8°, 46.2° and 67.5°. From the line broadening, an average size of the metal nanoparticles of 5.4 nm was calculated. For Ru/K1221 a very large broadening of the signal at 2θ value of 44.6° indicates significantly smaller metal particles. Similarly, for Ni/K1221 a very large broadening of the signal at 2θ value of 43.1° indicates the formation of very small metal particles. Additionally, a weak signal for NiO was observed indicating some oxidation of the metal nanoparticles. Thus, the size of the metal nanoparticles determined by XRD analysis is in good agreement with the size of the pores of water-swollen K1221.
 | ||
| Fig. 3 Powder X-ray diffraction analysis of Rh/K1221 (5.3%w/w Rh). | ||
Analysis of the particle distribution of Rh/K1221 by scanning electron microscopy revealed an even distribution of relatively small areas of high metal concentration throughout the polymer matrix (Fig. 4). EDX analysis showed that the metal was segregated in regions a few hundred of nanometres wide. It is clear, however, that the distribution of the metal in each of these regions is uneven (see the lower-right inset of Fig. 4). Moreover, XRD analysis reveals that the size of the metal nanoparticles is 4.7 nm. No sharp signals that could be attributed to larger metal particles were detected. This suggests that the Rh nanoparticles with mean diameter of 4.7 nm are formed preferentially in discrete domains of the polymer matrix, which are more or less evenly distributed throughout the entire volume of the beads. The existence of these domains where the metal tends to be accumulated is the likely consequence of structural characteristics of the sulfonated resin.
 | ||
| Fig. 4 SEM micrograph of the inside of a cut Rh/K1221 bead; the inserts show an EDS and an EDX image of the embedded Rh nanoparticles (right, top and bottom, respectively). | ||
Nitrobenzene hydrogenation
The ion-exchange resin K1221 with nanoparticles of four different metals (Rh, Pt, Ru, and Ni) were then tested in the hydrogenation of nitrobenzene as a representative consecutive reaction (Table 2).37,41–45 Intermediates (aniline) and products (cyclohexylamine) vary considerably in their properties (Scheme 2) and, thus, interact differently with the catalyst. Whereas nitrobenzene is a highly polar aromatic molecule (dipole moment 4.03 debye),46,47 the intermediate aniline is a less polar aromatic molecule (1.59 debye)46 and cyclohexylamine is an aliphatic molecule with even lower polarity (1.33 debye).48 Consequently, the average polarity of the reaction medium changes substantially during the hydrogenation reaction.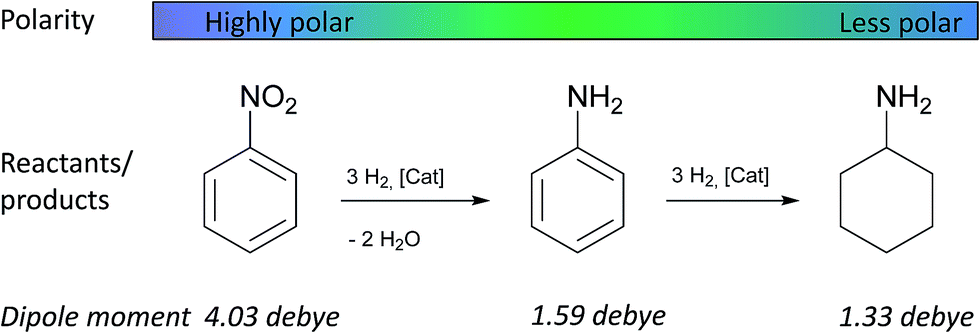 | ||
| Scheme 2 Hydrogenation of nitrobenzene to aniline and cyclohexylamine used as model of a consecutive reaction where the polarity of the reaction medium changes substantially during the reaction. | ||
The kinetics of the hydrogenation of nitrobenzene catalysts were explored for Rh/K1221, Pt/K1221, Ru/K1221, and Ni/K1221. Unexpectedly, the time-concentration profile was nearly identical for all four catalysts (Fig. 5 for Rh/K1221). Three distinct reaction phases were observed. In the first phase, the reaction was fast, whereby about 40% conversion was obtained in 12 min. This part of the profile is best described as a first-order reaction for nitrobenzene; the initial rate of reaction was −1.6 × 103 molNB molM−1 h−1 for Rh/K1221 (Table 1). Thereafter, in a second phase, the reaction stopped nearly entirely for 50 min and then started again, albeit at a much lower rate best described by a second-order reaction model. Both products aniline and cyclohexylamine appear to be formed in parallel (vide infra), while hardly any condensation (diphenylamine, N-cyclohexylaniline, dicyclohexylamine) and substitution products (cyclohexanol) arise.
 | ||
| Fig. 5 Time-concentration profile of the hydrogenation of nitro-benzene over Rh/K1221 (120 °C, 100 bar, 500 s m−1, THF/MeOH 9:1) showing the three distinct phases of the reaction in comparison to the profile of nitrobenzene hydrogenation over Rh/C as benchmark catalyst (insert); dicyclohexylamine (DHA) was formed only over Rh/C; trace amounts of diphenylamine (DPA), N-cyclohexylaniline (N-CHA), and cyclohexanol (CHN) were detected. | ||
To understand this unusual performance, the time-concentration profiles of the corresponding carbon-supported catalysts were recorded under the same conditions. Depending on the metal applied, remarkable differences were found (Table 1).44 Using Rh/C, the conversion of nitrobenzene proceeded within 12 min to provide a mixture of aniline, cyclohexylamine and dicyclohexylamine in a ratio of 80:19:1 (Fig. 2, insert). Once formed, the aromatic ring of aniline was not hydrogenated any further. Over Pt/C, nitrobenzene was converted after an initiation period to a mixture of aniline and cyclohexylamine in a ratio of 13:1.44 Over Ru/K1221 nitrobenzene was converted with a similar initial rate as over Ru/C. However, the rate slowed down over Ru/C at higher conversions and the formed aniline was converted to a mixture of cyclohexylamine and dicyclohexylamine. Over Ni/C, a short initiation period was observed. Then, nitrobenzene was first converted at a rate of −2.7 × 103 molNB molM−1 h−1, but then the reaction slowed down at higher conversions.44,49 Whereas aniline was formed as primary product, cyclohexylamine was formed in small amounts as a secondary product.
The similar time-concentration profiles observed for the polymer-supported catalysts clearly show that the reaction must have been governed by the (same) K1221 polymer support and not by the choice of the metal. Only with the polymer-supported nanoparticles did the reaction stop entirely after 12 min and eventually resume after 75 min (Fig. 2). This implies that the reaction occurred under a diffusion regime and that the unusual kinetic profile is related to features of the polymer support. Noteworthy is that all polymer-supported catalysts were selective to primary amines, while formation of the secondary amine dicyclohexylamine was observed over the classic carbon-supported catalysts (Rh/C and Ru/C). Moreover, a comparison of the polymer-supported catalysts (Ru/K1221, Ni/K1221) and the classic benchmark catalysts (Ru/C, Ni/C) revealed the absence of an initiation period. This suggests that the metal nanoparticles are protected against contact with air within the polymer matrix. These findings (diffusion regime, enhanced selectivity, protection of the metal against oxidation) suggest that the reactions take place inside the swollen polymeric framework. It has been reported that swelling of the gel-type resin K1221 leads to formation of pores in between the polymer chains with a diameter of 2–8 nm.50–53 Access to catalytic sites located inside of such an essentially microporous support brings about a kind of shape selectivity; in fact, the larger condensation products did not form over the polymer-supported catalysts.
In order to investigate whether a synergistic effect of two differently supported metals can be exploited to tune the catalytic activity and the selectivity, Pt/K1221 was tested in combination with Ni or Rh on K1221, Ru/K1221 with Ni or Rh or Pt on K1221, and Ni/K1221 with Rh on K1221. The concentration profiles revealed the same three different phases as identified when a single supported metal was used.‡ Similarly, polymer-supported alloys of Rh/Ni and Pt/Ru on K1221 were tested in the hydrogenation of the nitrobenzene. The concentration profile also showed the three distinct reaction phases. Furthermore, comparable percentages of conversion and selectivity towards the products were achieved.‡ This confirms that the reaction must have been governed by a diffusion mechanism which is controlled by the K1221 polymer support independent of the type of metal and the size of the nanoparticles.
Solvent uptake into the polymer beads
In the dry state, gel-type resins do not present any porosity and the beads are fully in a glass-like state. Only when soaked with a proper solvent, the nanocavities of the resin become accessible and a high specific surface is acquired.54 For the parent K1221 resin, the ISEC analysis of the water-swollen beads revealed a surface area of 2736 m2 g−1. The swelling ability is strongly dependent on the medium. To unravel this effect, the uptake of reactants, products and solvents into the polymer beads was measured (Table 3) as the Specific Absorbed Volume (SAV).55 SAV is the volume of a liquid sorbed by a solid upon contact. In the case of resins like K1221, it represents the swelling volume, i.e. the volume of liquid filling the pores of the swollen polymer framework. Some bias can arise from small amounts of liquid retained at the external surface of the polymer beads or in the inter-particle voids of the packed solid. This is not generally a problem, especially for comparative purposes in a data set, provided that the external liquid is drained off by filtration or centrifugation.| Guest moleculea | H-bond donor | Dipole moment μ [Debye] | Dielectric constant ε [−] | Up-take [ml g−1] |
|---|---|---|---|---|
| a NB: nitrobenzene; THF: tetrahydrofuran; MeOH: methanol; iPrOH: iso-propanol; ANI: aniline; CHA: cyclohexylamine.b Suspension in n-heptane.c Ratio (9:1) dipole moments and dielectric constants from ref. 44, 46 and 54–56; s.a. see above. |
||||
| —b | — | — | — | — |
| NB | No | 4.03 | 34.8 | 0.15 |
| THF | No | 1.75 | 7.58 | 0.07 |
| MeOH | Yes | 2.87 | 32.70 | 0.40 |
| Water | Yes | 1.85 | 78.3 | 0.67 |
| iPrOH | Yes | 1.66 | 19.92 | 0.23 |
| ANI | Yes | 1.59 | 6.9 | 2.22 |
| CHA | Yes | 1.33 | 2.0–2.2 | 0.80 |
| THF/MeOHc | Mixture | s.a. | s.a. | 0.16 |
While the uptake was relatively low for the most polar (0.15 ml g−1 for nitrobenzene) and non-polar molecules (0.07 ml g−1 for THF) in the reaction mixture, it was significantly higher for molecules able to form hydrogen bonds (2.22, 0.80 and 0.40 ml g−1 for aniline, cyclohexylamine and methanol, respectively). Most likely, the high propensity of the beads of K1221 to absorb molecules with hydrogen bonding ability is related to the polar sulphonate groups present along the polymer chains. The use of a mixture of THF/MeOH (9:1) led to a significantly higher uptake (0.16 ml g−1) in comparison to neat THF, suggesting preferential uptake of methanol into the pores. Such selective sorption in cross-linked polymers from mixtures of miscible liquids is a well-known phenomenon.56,59 The solvent uptake goes along with a more or less large increase in the physical radius of the polymer beads (from 261(5) to 288–315 μm diameter),58 which implies, at the microscopic level, an increase in the average distance between the polymer chains. Hence, with these polymer-supported catalysts the course of the reaction will be affected by the swelling behaviour of the polymer in dependence of the composition of the reaction medium.60–62 For this reason macroreticular resins, whose behaviour is less dependent on swelling, are most often employed in industrial applications.61
Regarding the reaction, it was observed that it temporarily stops as soon as the concentration of aniline has reached a critical value corresponding to roughly 40% conversion of nitrobenzene. The build-up of aniline ought to have an effect on the polymer beads due to the strong absorption of aniline into the K1221 beads. Moreover, it is formed within the polymer framework and does not need to diffuse from outside to start interacting with the polymer framework. The large preference of K1221 for the absorption of aniline suggests that when it is formed in high enough amounts, the diffusion of compounds with much smaller swelling influence on K1221 from the outside is stopped. When large amounts of aniline are present, nitrobenzene and other species that have a low swelling power on K1221 may even be displaced from the support. Occasionally, we observed an increase in the concentration of nitrobenzene and decrease in the aniline concentration in the liquid phase at the transition from phase I to phase II (see the profile for Ru/K1221 in Fig. S7, ESI‡). Thus, a first consequence of the build-up of aniline in the catalyst is a shortage of the substrate within the catalyst, which readily explains why the formation of aniline stops at a sufficiently high conversion of nitrobenzene.
Yet, something else must have occurred, as also the formation of cyclohexylamine stops during phase II and, later on, the catalytic activity is restored in phase III. The build-up of aniline in the catalyst brings its local concentration nearby the metal nanoparticles to relatively high levels. As shown schematically in Schemes 1 and 3, the metal nanoparticles within the polymer network of the support are surrounded by the polymer chains. During phase I the polymer framework is swollen by the solution of NB in THF/MeOH. These agents are characterised by a relatively small uptake, hence the swelling degree of the polymer will be small as well. This situation is schematically depicted in the upper part of Scheme 3. Under these conditions there is little room, due to the steric hindrance of the polymer chains, for the side-on adsorption of aniline. Moreover, the phenyl rings of the styrene units of K1221 likely compete with aniline for the η6 adsorption over the active surface. In this context, soluble polystyrene has been showed to be an effective stabilizer for Sc, Os, Pd, and Ru,63 Au64 and Pt65 through the interaction of the π electrons of the phenyl rings with the unsaturated surface metal atoms. Side-on adsorption is necessary for activation of the phenyl ring of aniline and is a condition for hydrogenation of the aromatic ring.45,49,66 As side-on adsorption is not necessary for the hydrogenation of nitrobenzene, this reaction proceeds smoothly.
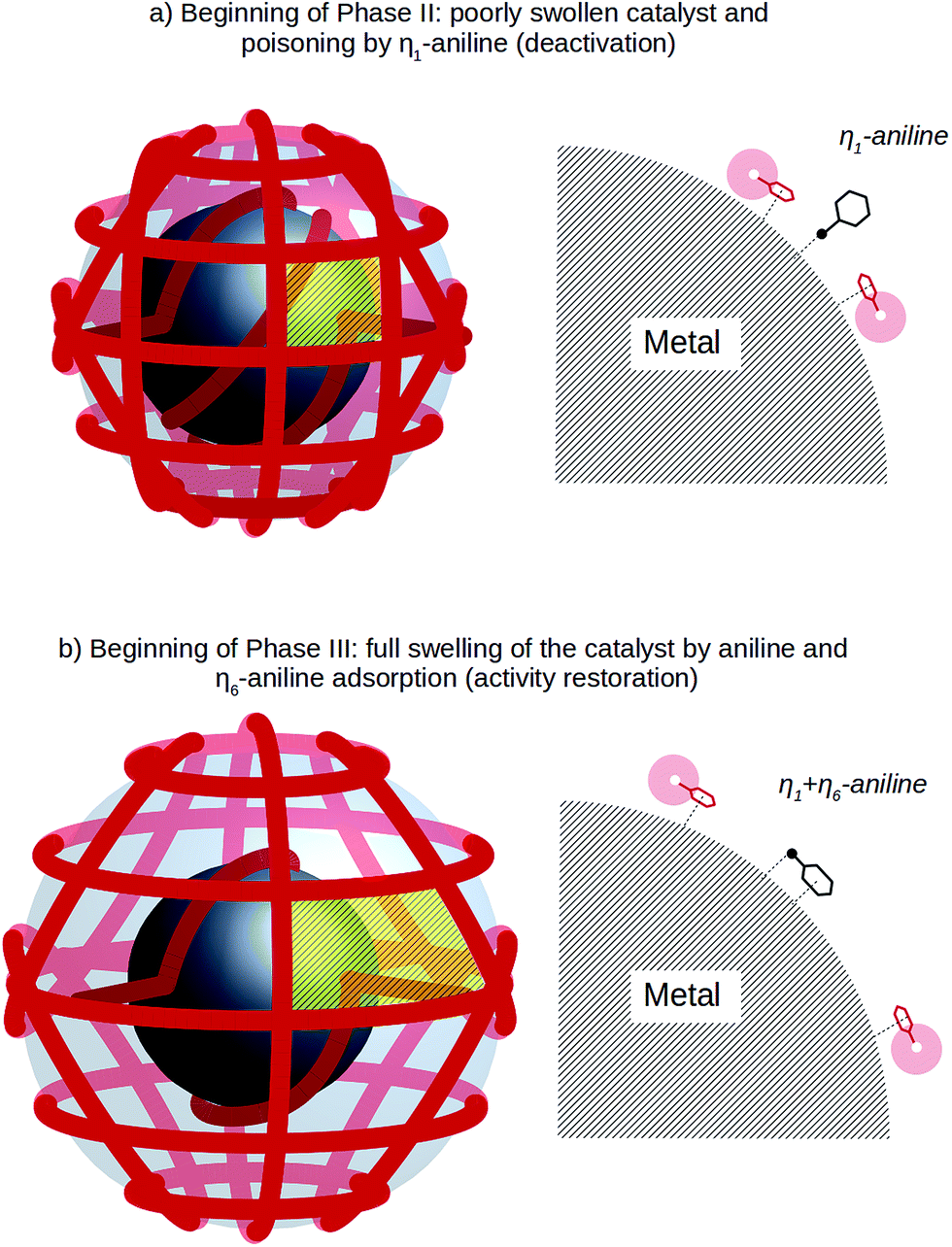 | ||
| Scheme 3 Schematic representation of the adsorption mode of aniline on the surface of a metal nanoparticle in dependence of the competitive adsorption of the phenyl groups of the K1221 polymer support changing with the degree of swelling (low in the upper; higher in the lower part). | ||
When the aniline concentration inside the catalyst reaches a critical value, its η6 adsorption is so unfavourable that hydrogenation of aniline to cyclohexylamine67 is suppressed. In effect, the catalyst is poisoned by the phenyl groups of the polymer and by η1-adsorbed aniline, which also stops the hydrogenation of nitrobenzene.
However, the build-up of aniline has also a second, favourable effect. When it is present in relatively high amounts, such as at the beginning of phase II, the polymer starts to swell. The expansion of the polymer framework, which is represented in the lower part of Scheme 3, pulls the polymeric chains away from the metal nanoparticles so that there is more room for the aniline molecules to tilt from end-on to side-on adsorption on the metal surface. Moreover the higher distance of the polymeric chains from the metal surface makes their interaction more difficult. In consequence, the conditions which caused the catalyst to stop working at the end of phase I are progressively removed during phase II, so that the catalytic activity is resumed in phase III.
However the steric constraint, although relieved at the beginning of phase III in comparison with the beginning of phase II, can still give rise to a shape-selectivity effect preventing the formation of the condensation products too bulky to fit the room available around the metal nanoparticles.
Conclusions
Unlike oxidic supports, polymer supports are highly flexible materials. In the presence of suitable solvents, the pores of the polymer support open to allow for chemical reactions and close when the beads are removed from the solvent. The dimensions of the solvent network formed in the swollen state control the maximum size of metal nanoparticles generated during the synthesis of polymer-supported catalysts. In the dry state, the metal nanoparticles are perfectly trapped in the polymer matrix. As the nanoparticles are then encapsulated in the polymer matrix, they are well protected against contact with air. Dispersed in a solvent, the resin support shows highly dynamic behaviour, whereby the accessibility of the porous network changes with the polarity and nature of the medium. The substrate molecules diffuse through the solvent network to access the catalytic sites on the surface of the metal nanoparticles. Consequently, the dynamic properties of the polymeric support strongly influence the catalytic activity.Here, the sulphonated gel-type ion-exchange resin K1221 was shown to be a versatile support for several metals. During the hydrogenation of nitrobenzene to cyclohexylamine, the very high affinity of the intermediate aniline towards the polymeric support, explains why the reaction, after proceeding at a high rate during the first 12 min, completely stops. In fact, build-up of aniline within the polymer framework leads to progressive swelling of the support and exclusion of the substrate from the catalysts. Slow relief of the steric hindrance at a later stage allows the catalytic activity to be restored. Moreover, the tight polymer network around the metal nanoparticles controls the selectivity preventing the formation of condensation products by a shape-selectivity effect.
Accordingly, the polymeric support gives a performance completely different from that of inorganic supports of classic heterogeneous metal catalysts. On the one hand, the confinement of the metal nanoparticles within the polymer framework of a gel-type resin makes the catalysts work under a diffusion regime and confers to the support the full control of the catalytic reaction. On the other hand, the steric effects of the confinement are modulated by the swelling of the polymeric support. If properly controlled, this dynamic behaviour can be utilized to switch the activity of the encapsulated metal nanoparticles on and off at a certain status of the reaction. This gives control to catalytic activity and selectivity at a specific time of the reaction.
Thus, the flexible matrix of gel-type polymeric supports enables developing smart catalysts based on immobilized metal nanoparticles.
Acknowledgements
This work originated from an exchange of students between Universita' degli Studi di Padova and RWTH Aachen University. We acknowledge the support of both universities. Also we are grateful to Elise Keitel (IME) for the elemental analyses. We are also highly grateful to Sabrina Mallmann and Rahimi Khosrow from DWI for XRD and SEM analysis.Notes and references
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18 CrossRef CAS PubMed.
- A. Drelinkiewicz and M. Hasik, J. Mol. Catal., 2001, 177, 149 CrossRef CAS.
- C. Burato, P. Centomo, M. Rizzoli, A. Biffis, S. Campestrini and B. Corain, Adv. Synth. Catal., 2006, 348, 255 CrossRef CAS PubMed.
- M. Králik and A. Biffis, J. Mol. Catal. A: Chem., 2001, 177, 113 CrossRef.
- B. Corain and M. Kralik, J. Mol. Catal. A: Chem., 2001, 173, 99 CrossRef CAS.
- C. M. Park, M. S. Kwon and J. Park, Synthesis, 2006, 22, 3790 CrossRef PubMed.
- R. Nakao, H. Rhee and Y. Uozumi, Org. Lett., 2005, 7, 163 CrossRef CAS PubMed.
- M. Hronec, Z. CvengroSov, M. Krhlik, G. Palma and B. Corain, J. Mol. Catal., 1996, 105, 25 CrossRef CAS.
- M. Liu, M. Han and W. W. Yu, Environ. Sci. Technol., 2009, 43, 2519 CrossRef CAS.
- M. Liua, H. Xiaoa, H. Shanb, X. Lia, Y. Liua and A. Wang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 699 CrossRef PubMed.
- L. M. Bronstein, G. Goerigk, M. Kostylev, M. Pink, I. A. Khotina, P. M. Valetsky, V. G. Matveeva, E. M. Sulman, M. G. Sulman, A. V. Bykov, N. V. Lakina and R. J. Spontak, J. Phys. Chem., 2004, 108, 18234 CrossRef CAS.
- S. Sakar, E. Guibal, F. Quignard and A. K. SenGupta, J. Nanopart. Res., 2012, 14, 715 CrossRef.
- G. Schmidt, N. Toshima and B. Corrain, Metal Nanoclusters in Catalysis and Materials Science: The Issue of Size Control, 1st edn, Elsevier, 2007 Search PubMed.
- L. Cumbal, J. Greenleaf, D. Leun and A. K. SenGupta, React. Funct. Polym., 2003, 54, 167 CrossRef CAS.
- S. Sarkar, E. Guibal, F. Quignard and A. K. SenGupta, J. Nanopart. Res., 2012, 14, 715 CrossRef.
- P. Barbaro and F. Liguori, Platinum Met. Rev., 2011, 55, 180 CrossRef.
- S. Alabbad, S. F. Adil, M. E. Assal, M. Khan, A. Alwarthan and M. R. Siddiqui, Arabian J. Chem., 2014, 7, 1192 CrossRef CAS PubMed.
- P. D. Stevens, J. Fan, H. M. R. Gardimalla, M. Yen and Y. Gao, Org. Lett., 2005, 7, 2085 CrossRef CAS PubMed.
- J. Govan and Y. K. Gun'ko, Nanomaterials, 2014, 4, 222 CrossRef CAS PubMed.
- S. Doherty, J. G. Knight, J. R. Ellison, D. Weekes, R. W. Harrington, C. Hardacre and H. Manyar, Green Chem., 2012, 14, 925 RSC.
- M. Rose, ChemCatChem, 2014, 6, 1166 CAS.
- M. M. Dell'Anna, G. Romanazzi and P. Mastrorilli, Curr. Org. Chem., 2013, 17, 1236 CrossRef.
- M. L. Richards and P. J. H. Scott, in Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. W. Cue, John Wiley & Sons Ltd., Chichester, 2012, p. 185 Search PubMed.
- R. Liu, C. Sosa, Y.-W. Yeh, F. Qu, N. Yao, R. K. Prud'homme and R. D. Priestley, J. Mater. Chem. A, 2014, 2, 17286 CAS.
- K. R. Reddy, K.-P. Lee and A. I. Gopalan, J. Nanosci. Nanotechnol., 2007, 7, 3117 CrossRef CAS PubMed.
- K. R. Reddy, K.-P. Lee, A. I. Gopalan, M. S. Kim, A. M. Showkat and Y. C. Nho, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3355 CrossRef CAS PubMed.
- K. L. Luska, P. Migowski and W. Leitner, Green Chem., 2015, 17(6), 3195 RSC.
- Z. Hou, N. Theyssen, A. Brinkmann and W. Leitner, Angew. Chem., Int. Ed., 2005, 44(9), 1346 CrossRef CAS PubMed.
- A. Kraynov and T. E. Müller, in Applications of Ionic Liquids in Science and Technology, ed. S. Handy, InTech, Rijeka, 2011 Search PubMed.
- B. Corain, M. Zecca and K. Jeřábek, J. Mol. Catal. A: Chem., 2001, 177, 3 CrossRef CAS.
- Z. You, T. Minami, C. Yin, Y. Umehara, T. Matsumura, C. Hashimoto and Y. Hosono, US pat., vol. US8802893 B2, 2014.
- E. V. d. Steene, J. D. Clercq and J. W. Thybaut, Chem. Eng. J., 2014, 242, 170 CrossRef PubMed.
- M. Iwahara, vol. US 2003/0013925 A1, 2003.
- B. Corain, K. Jerabek, P. Centomo and P. Canton, Angew. Chem., Int. Ed., 2004, 43, 959 CrossRef CAS PubMed.
- B. Corain, M. Zecca, P. Canton and P. Centomo, Philos. Trans. R. Soc., A, 2010, 368, 1495 CrossRef CAS PubMed.
- R. Knapp, S. A. Wyrzgol, M. Reichelt, T. Hammer, H. Morgner, T. E. Müller and J. A. Lercher, J. Phys. Chem. C, 2010, 114, 13722 CAS.
- M. Kralik, R. Fisera, M. Zecca, A. A. D'Archivio, L. Galantini, K. Jerabek and B. Corain, Collect. Czech. Chem. Commun., 1998, 63, 1074 CrossRef CAS.
- K. Jeřábek, Anal. Chem., 1985, 57, 1595 CrossRef.
- K. Jeřábek, Anal. Chem., 1985, 57, 1598 CrossRef.
- P. Centomo, P. Canton, M. Ferroni and M. Zecca, New J. Chem., 2010, 34, 2956 RSC.
- P. Centomo, M. Zecca, M. Kralik, D. Gasparovicova, K. Jerabek, P. Canton and B. Corain, J. Mol. Catal. A: Chem., 2009, 300, 48 CrossRef CAS PubMed.
- R. Fisera, M. Kralik, J. Annus, V. Kratky, M. Zecca and M. Hronec, Collect. Czech. Chem. Commun., 1997, 62, 1763 CrossRef CAS.
- M. Kralik, B. Corain and M. Zecca, Chem. Pap., 2000, 54, 254 CAS.
- P. Tomkins, E. Gebauer-Henke, W. Leitner and T. E. Müller, ACS Catal., 2015, 5, 203 CrossRef CAS.
- E. Gebauer-Henke, W. Leitner, A. Prokofieva, H. Vogt and T. E. Müller, Catal. Sci. Technol., 2012, 2, 2539 CAS.
- T. R. Griffiths and D. C. Pugh, Coord. Chem. Rev., 1979, 29, 129 CrossRef CAS.
- M. Dutkiewicz, B. Szurkowski and T. Hilczer, Chem. Phys. Lett., 1983, 94, 531 CrossRef CAS.
- M. D. Gregory, H. E. Affsprung and S. D. Christian, J. Phys. Chem., 1967, 1748 Search PubMed.
- E. Gebauer-Henke, P. Tomkins, W. Leitner and T. E. Müller, ChemCatChem, 2014, 6, 2910 CrossRef CAS PubMed.
- K. Jeřábek, in Cross Evaluation of Strategies in Size-Exclusion Chromatography, ed. M. Potschka and P. L. Dubin, American Chemical Society, Washington DC, 1996, vol. 635, ACS Symposium Series, p. 211 Search PubMed.
- B. Corain, M. Zecca, P. Canton and P. Centomo, Philos. Trans. R. Soc., A, 2010, 368, 1495 CrossRef CAS PubMed.
- B. Corain, P. Centomo, C. Burato and P. Canton, in Metal Nanoclusters in Catalysis and Material Science. The issue of the size control, Elsevier, 2011, p. 413 Search PubMed.
- B. Corain, K. Jerabek, P. Centomo and P. Canton, Angew. Chem., Int. Ed., 2004, 43, 959 CrossRef CAS PubMed.
- B. Corain and M. Kralik, J. Mol. Catal. A: Chem., 2000, 159, 153 CrossRef CAS.
- K. W. Pepper, D. Reichenberg and D. K. Hale, J. Chem. Soc., 1952, 3129 RSC.
- D. C. Sherrington, in Polymer-supported Reactions in Organic Synthesis, ed. P. Hodge and D. C. Sherrington, Wiley, New York, 1980, p. 1 Search PubMed.
- I. Fischer, Acta Chem. Scand., 1950, 4, 1197 CrossRef CAS PubMed.
- P. Centomo, K. Jeřábek, D. Canova, A. Zoleo, A. L. Maniero, A. Sassi, P. Canton, B. Corain and M. Zecca, Chem.–Eur. J., 2012, 18, 6632 CrossRef CAS PubMed.
- M. Zecca, R. Fišera, G. Palma, S. Lora, M. Hronec and M. Kralik, Chem.–Eur. J., 2000, 6, 1980 CrossRef CAS.
- K. Jeřábek, L. Hanková and L. Holub, J. Mol. Catal. A: Chem., 2010, 333, 109 CrossRef PubMed.
- R. Banavali, R. T. Hanlon, K. Jeřábek and A. K. Schultz, in Catalysis of organic reactions: twenty-second conference (chemical industries), ed. P. ML, CRC, New York, 2008, p. 279 Search PubMed.
- P. Centomo, I. Bonato, L. Hanková, L. Holub, K. Jeřábek and M. Zecca, Top. Catal., 2013, 56, 611 CrossRef CAS PubMed.
- S. Kobayashi and R. Akiyama, Chem. Commun., 2003, 449 RSC.
- H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 4151 CrossRef CAS PubMed.
- A. Ohtaka, Y. Kono, S. Inui, S. Yamamoto, T. Ushiyama, O. Shimomura and R. Nomura, J. Mol. Catal. A: Chem., 2012, 360, 48 CrossRef CAS PubMed.
- P. Schärringer, T. E. Müller, A. Jentys and J. A. Lercher, J. Catal., 2009, 263, 34 CrossRef PubMed.
- E. A. Gelder, Chem. Commun., 2005, 522 RSC.
Footnotes |
| † In memory of the late Professor Benedetto Corain (July 8th 1941 – September 24th 2014), twice Humboldt-Stipendiat, enthusiastic man and tireless researcher. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05061h |
| This journal is © The Royal Society of Chemistry 2015 |