
Re2O7 catalyzed dienone-phenol rearrangement†
Abstract
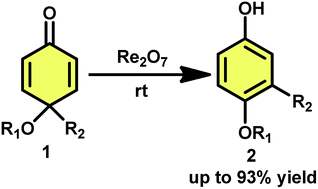
The dienone-phenol rearrangement of 4,4-disubstituted cyclohexadienones catalyzed by Re2O7 has been described. Multi-substituted phenols can be efficiently obtained in good to excellent yields by employing this catalytic protocol.
 Please wait while we load your content...
Please wait while we load your content...

