
A review on recent developments of anion exchange membranes for fuel cells and redox flow batteries
Abstract
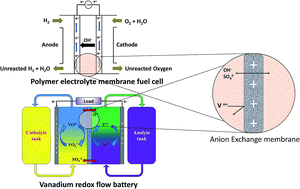
Cation exchange membranes (CEMs) have attracted tremendous attention in electrochemical energy conversion and storage systems owing to their high proton conductivity and chemical stability. However, applications of CEMs suffer from a number of disadvantages such as requirement of costly platinum catalyst, and high crossover of fuels or positively charged redox species due to the electro-osmotic drag. Anion exchange membranes (AEMs) have shown promising characteristics to overcome some of the problems associated with CEMs; the advantages of AEMs being selective transport anionic charge carriers, lower crossover of cationic redox couples, and facile reaction kinetics in energy conversion processes. These unique properties of AEMs result mainly from the density and distribution of positively charged functional groups, along with a macromolecular polymer backbone. As a result, there has been an increasing demand for the development of AEMs with better selectivity, higher chemical stability and conductivity, and a lot of work has been carried out in this area. The aim of this review is to discuss developments in the synthesis and applications of AEMs in the field of electrochemical energy conversion and storage, on which many researchers have been working in recent years.
 Please wait while we load your content...
Please wait while we load your content...

