DOI:
10.1039/C5RA03333K
(Review Article)
RSC Adv., 2015,
5, 37231-37274
Activation of C–H bonds of thiosemicarbazones by transition metals: synthesis, structures and importance of cyclometallated compounds
Received
23rd February 2015
, Accepted 24th March 2015
First published on 24th March 2015
Abstract
Transition metals, namely, palladium(II), platinum(II), ruthenium(II), rhodium(III) and iridium(III) have induced activation of C–H bonds of thiosemicarbazones and yielded mono-, di-, tri- and tetra-nuclear complexes with or without tertiary phosphines as co-ligands. Mono-thiosemicarbazones (R1R2C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N3–N2H–C1(S)–N1R3R4) undergo loss of C–H (R1 ring) and N2–H protons and formed cyclometallated derivatives. In mono- and di-nuclear complexes, the thio-ligands coordinate as terdentate (C,N,S) dianions, while in tri- and tetra-nuclear complexes, these ligands act as tetradentate (C,N,μ-S) dianions. The mono-thiosemicarbazones generally involve μ-S bridging in oligomers. Only one bis-thiosemicarbazone is reported to form mononuclear or mixed metal di-nuclear complexes. This review describes synthetic aspects, molecular structures, electronic absorption spectroscopy, fluorescence, cyclic voltammetry, NMR(1H, 13C, 31P) spectroscopy and applications (biological, catalysis) of complexes. The factors for metallation and conclusions with future scope of investigations are also mentioned. Literature coverage is upto date, to the best of my knowledge, though a few omissions cannot be ruled out.
N3–N2H–C1(S)–N1R3R4) undergo loss of C–H (R1 ring) and N2–H protons and formed cyclometallated derivatives. In mono- and di-nuclear complexes, the thio-ligands coordinate as terdentate (C,N,S) dianions, while in tri- and tetra-nuclear complexes, these ligands act as tetradentate (C,N,μ-S) dianions. The mono-thiosemicarbazones generally involve μ-S bridging in oligomers. Only one bis-thiosemicarbazone is reported to form mononuclear or mixed metal di-nuclear complexes. This review describes synthetic aspects, molecular structures, electronic absorption spectroscopy, fluorescence, cyclic voltammetry, NMR(1H, 13C, 31P) spectroscopy and applications (biological, catalysis) of complexes. The factors for metallation and conclusions with future scope of investigations are also mentioned. Literature coverage is upto date, to the best of my knowledge, though a few omissions cannot be ruled out.
 Tarlok S. Lobana | Tarlok Singh Lobana born in 1950 in Gado Majra Village (Rajpura) in Punjab graduated in 1969 from S A College Ambala, post-graduated from Panjabi University, Patiala and received his Ph.D. degree from Guru Nanak Dev University (GNDU) in 1977. From the position of Research Assistant (1975), he rose to the level of Professor of Chemistry (1991) and retired in December 2010 and was reemployed until December 2012 and is Emeritus Scientist at GNDU with effect from January 2013 sponsored under Council of Scientific and Industrial Research New Delhi. His research interest have been coordination chemistry, pre-designed synthesis of metal derivatives of N,S-donors, metal mediated C–S rupture, organometallic chemistry. He has been Chairman of Department (May 1997 November 1999) and later Dean Sciences 2001 to 2002. He has been Fellow of National Academy of Sciences in 1999. He was the recipient of the Bronze Medal of the Chemical Research Society of India (2002). He has been INSA-Royal Society Fellow (UK, 1989), EPFL Visiting Professor Laussane (Suisse, 1991), DAAD Fellow (Munich, 1992), Spain Govt Visiting Fellow (1996–1997), JSPS Visiting Fellow (2002–2003), Visiting Professor at National Christian University Chungli Taiwan (2005). He has been complimentary e-member of Royal Society of Chemistry 2012 to 2014. He has published 215 research papers/reviews in international journals of repute. |
1. Introduction
The intramolecular activation of aromatic C–H bonds of coordinated ligands such as Schiff bases, azines etc. by transition metals represents an active area of research because cyclometallated compounds show significant applications, such as their use in regiospecific organic and organometallic synthesis, in insertion reactions, in the synthesis of new metal mesogenic compounds, reactions with nucleophiles and in catalytic materials, as liquid crystals, as analytical tools and for the design of metal complexes with promising anticancer or photochemical properties.1–23 The activation of C–H bonds of thiosemicarbazones with transition metals has also received some attention and the current review is intended to describe this area.
The coordination chemistry of thiosemicarbazones describing bonding, structural aspects, biological, catalytical and analytical applications has received attention of many investigators.24–26 This review begins with brief general introductory comments about thiosemicarbazones/their complexes highlighting their importance. The intramolecular activation of C–H bonds of aromatic, aliphatic or heterocyclic rings of coordinated thiosemicarbazones by the transition metals is surveyed and described in this review. The synthetic aspects, molecular structures, electronic absorption spectroscopy, fluorescence, cyclic voltammetry, NMR (1H, 13C, 31P) spectroscopy and applications (biological, catalysis) of cyclometallated complexes form the main focus of review. The factors for metallation and conclusions with future scope of investigations are also discussed.
2. Thiosemicarbazones – introductory comments
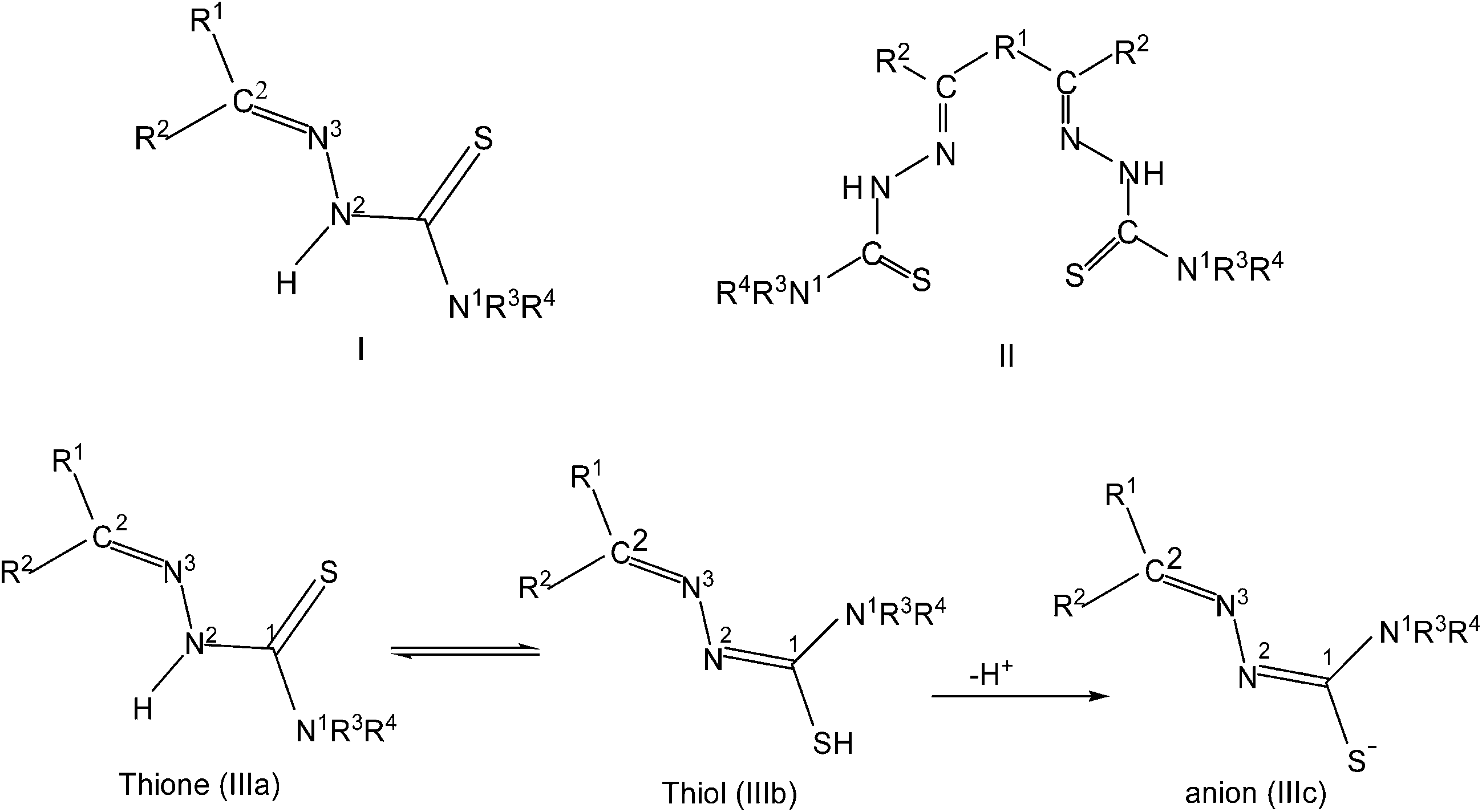
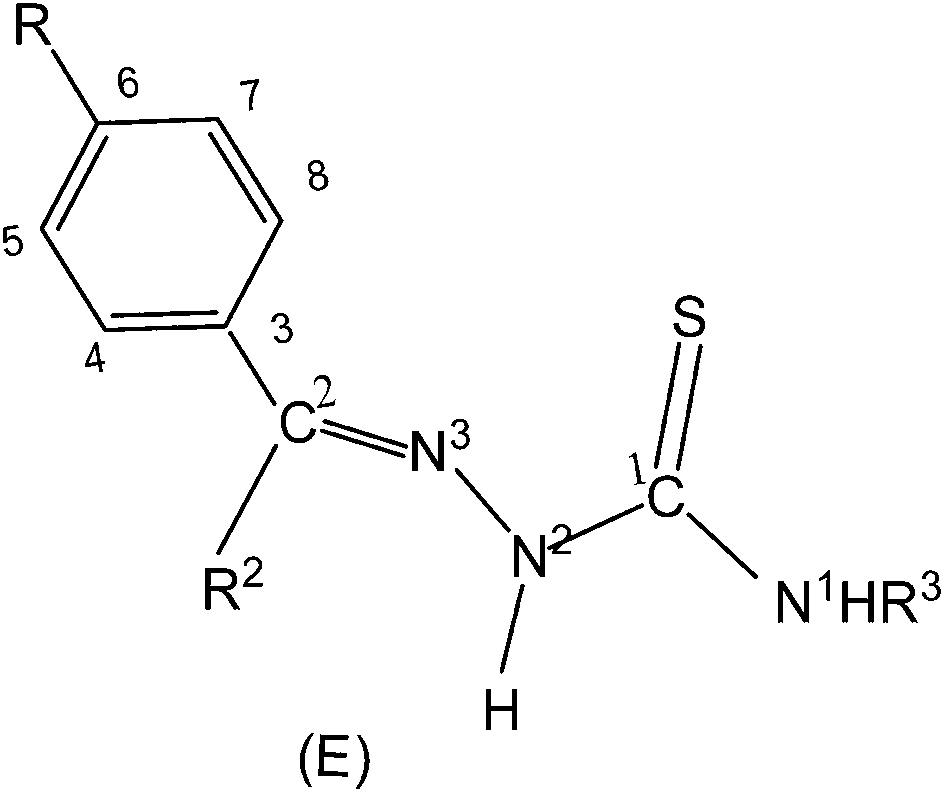
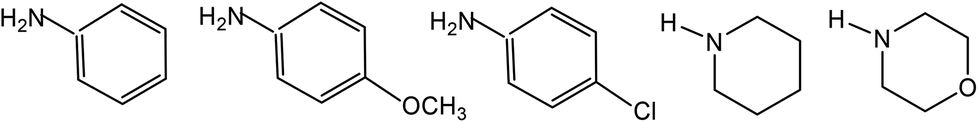
Thiosemicarbazones are normally N,S-donor ligands and represent a class of Schiff bases which can be obtained by the condensation of an aldehyde, or a ketone with a thiosemicarbazide (eqn (1)). Broadly speaking these thiosemicarbazones are either mono-thiosemicarbazones (R1R2CN–NH–C(S)–NR3R4, Structure I) or bis-thiosemicarbazones (Structure II). In mono-thiosemicarbazones, the substituents R1, R2, R3 and R4 could be different, and this provides a great possibility of variation of this class of thio-ligands. Likewise in bis-thiosemicarbazones two arms are connected via a ring, or a C–C bond.26| | |
R2CO + H2N–NH–C(S)–NH2 → R2CN–NH–C(S)–NH2
| (1) |
Mono-thiosemicarbazones exist as thione–thiol tautomers (IIIa–IIIb) and can bind to a metal center in the neutral (IIIa), or the anionic forms (IIIc). The form IIIc is generated after loss of –N2H or –SH hydrogen ions of a thiosemicarbazone and a large number of bonding modes have been observed in their metal complexes wherein a thio-ligand is coordinating in neutral or anionic forms.27–43 Thiosemicarbazones are potential chemotherapeutics and are noted for their pharmacological properties, particularly as antiparasitic,44–49 as antibacterial,50–52 and as antitumoral agents.53–56 Further, metal complexes of thiosemicarbazones have shown better biological activity than the parent thiosemicarbazones.26,57–68 Significantly, among various metals, palladium(II) and copper(II) complexes with thiosemicarbazones have been particularly interesting because of their antitumor, antibacterial, antifungal, catalytic activities and ion sensors.26,69–89
3. Metallation of thiosemicarbazones
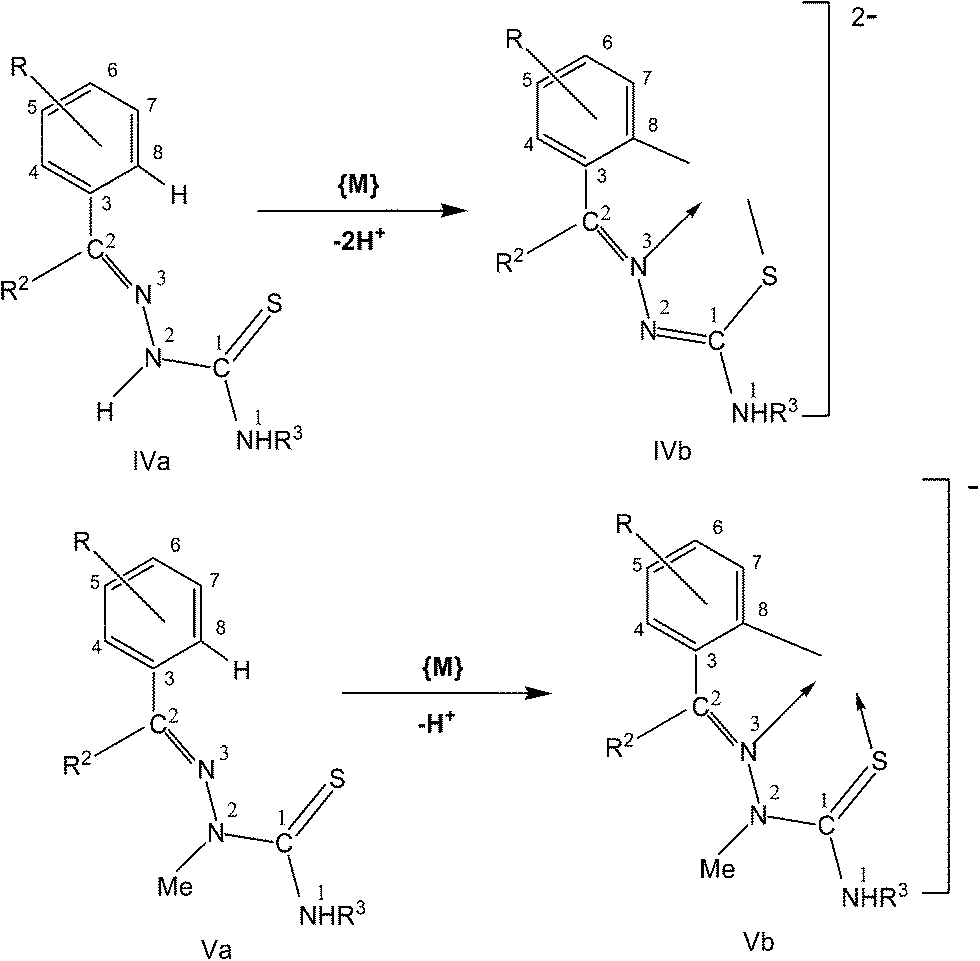
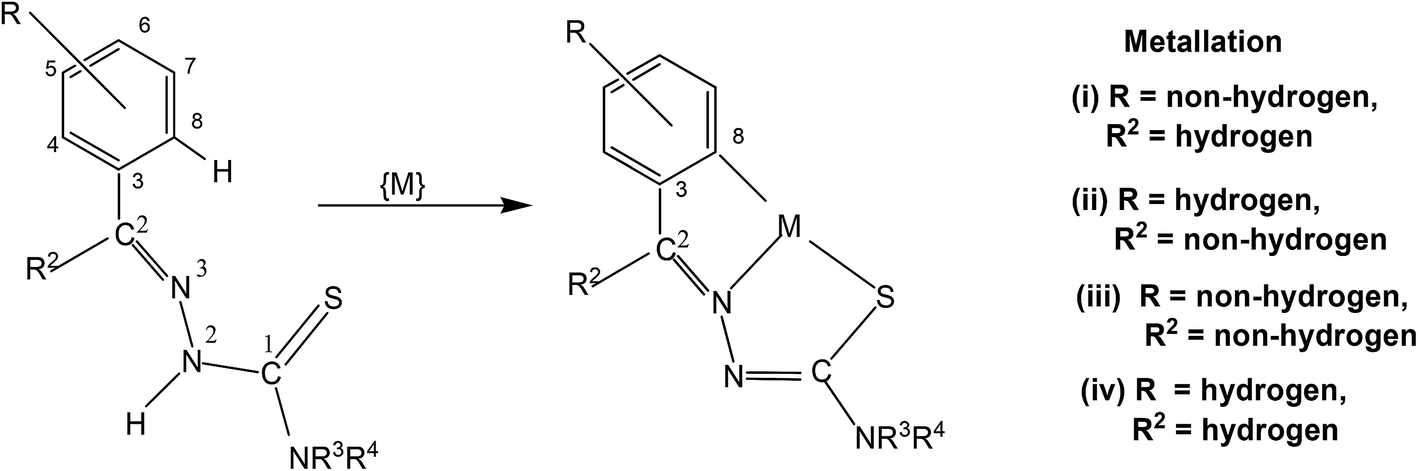
In this section, only such reaction systems are a focus of discussion which involve formation of metal–carbon bonds via activation of C–H bonds of rings (R1) of thiosemicarbazones (R1R2CN–NH–C(S)–NR3R4, Structure I). Various complexes formed via activation of C–H bonds, their structures, mechanistic pathways, factors promoting metallation, biological, catalytical and analytical aspects will be covered. Complexes having metal–carbon bonds which do not originate from thiosemicarbazones are excluded from this review. However, there are reviews reported in the literature, which cover M–C bonds formed by auxiliary alkyl or aryl groups with no activation of C–H bonds of thiosemicarbazones in forming M–C bonds.26,90–94 Reaction systems where activation was expected but did not occur may also find mention in the review where relevant. It is the mono-thiosemicarbazone ligands which have predominantly shown cyclometallation. There is only one report where a bis-thiosemicarbaone showed C–H activation. The subject matter under review is discussed under different sub-heads. The general structure of mono-thiosemicarbazones exhibiting metallation (Structure I) shall be used for defining various thiosemicarbazones with different substituents at C2, N2, and N1 atoms in the forthcoming discussion. Thiosmeicarbazones lose a ring proton (C–H) and –N2H proton and coordinate to the metal ions as terdentate (C,N,S) dianions involving μ-S bridging only in oligomers (Structure IVb), excepting a few cases where they coordinate as monoanion as shown in structure Vb. Bis-thiosemicarbazones have coordination pattern similar to IVb (mononuclear complexes) but use both arms (Structure II) in the same way for diuclear complexes. In that case they act as hexadenate tetra-anions. The subject matter of review is discussed under different headings: synthetic aspects, molecular structures, electronic absorption spectroscopy and fluorescence, cyclic voltammetry, NMR spectroscopy, factors promoting metallation, applications – biological and catalysis and finally conclusion.
Several complexes have co-ligands and thus various co-ligands which appear in this review are summarized here.
PPh3: triphenylphosphine;
MePh2P: methyldiphenylphosphine;
dppm: bis(diphenylphopshino)methane, Ph2P–CH2–PPh2
dppe: 1,2-bis(diphenylphophino)ethane, Ph2P–(CH2)2–PPh2
dppp: 1,3-bis(diphenylphophino)propane, Ph2P–(CH2)3–PPh2
dppb: 1,4-bis(diphenylphophino)butane, Ph2P–(CH2)4–PPh2
dppn: 1,5-bis(diphenylphophino)pentane, Ph2P–(CH2)5–PPh2
dpph: 1,6-bis(diphenylphophino)hexane, Ph2P–(CH2)6–PPh2
vdpp: 1,1-bis(diphenylphosphino)vinylene, CH2C(PPh3)2
dppen: cis- and trans-1,2-bis(diphenylphophino)ethene, Ph2P–CHCH–PPh2
bdpb: ortho-bis(diphenylphosphino)benzene, o-C6H4(PPh2)2
bdcp: bis(diphenylphosphino)cyclopropane, (CH2)2C(PPh3)2
Fc: 1,1′-bis(diphenylphosphino)ferrocene, PPh2–C5H4–Fe–C5H4–PPh2
3.1 Synthetic aspects
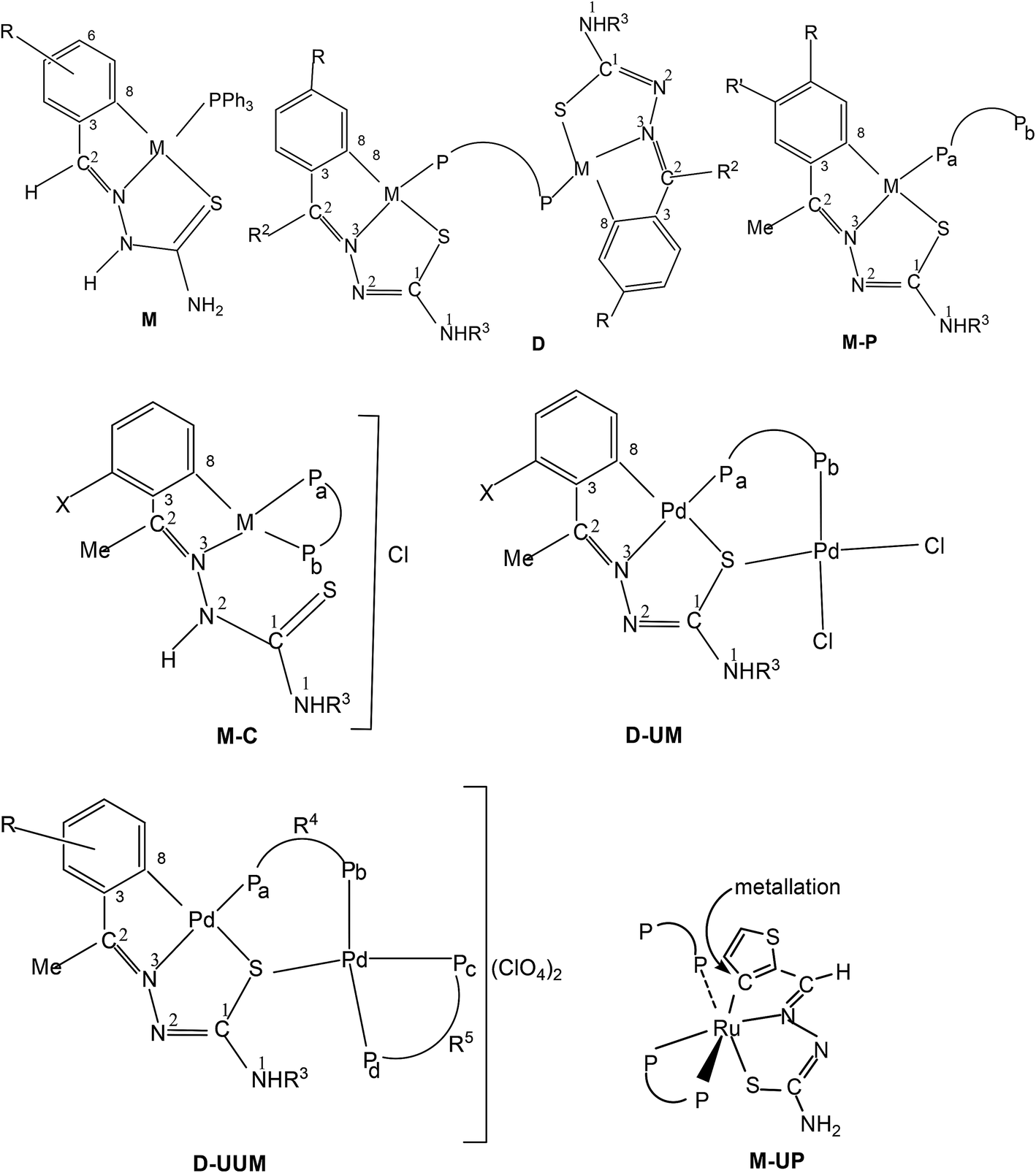
Palladium. Palladium(II) is the one single metal ion which has shown the largest efficiency in activation of C–H bonds of ring substituents of coordinated thiosemicarbazones. The details of complexes displaying activation of C–H bonds are described in this section.
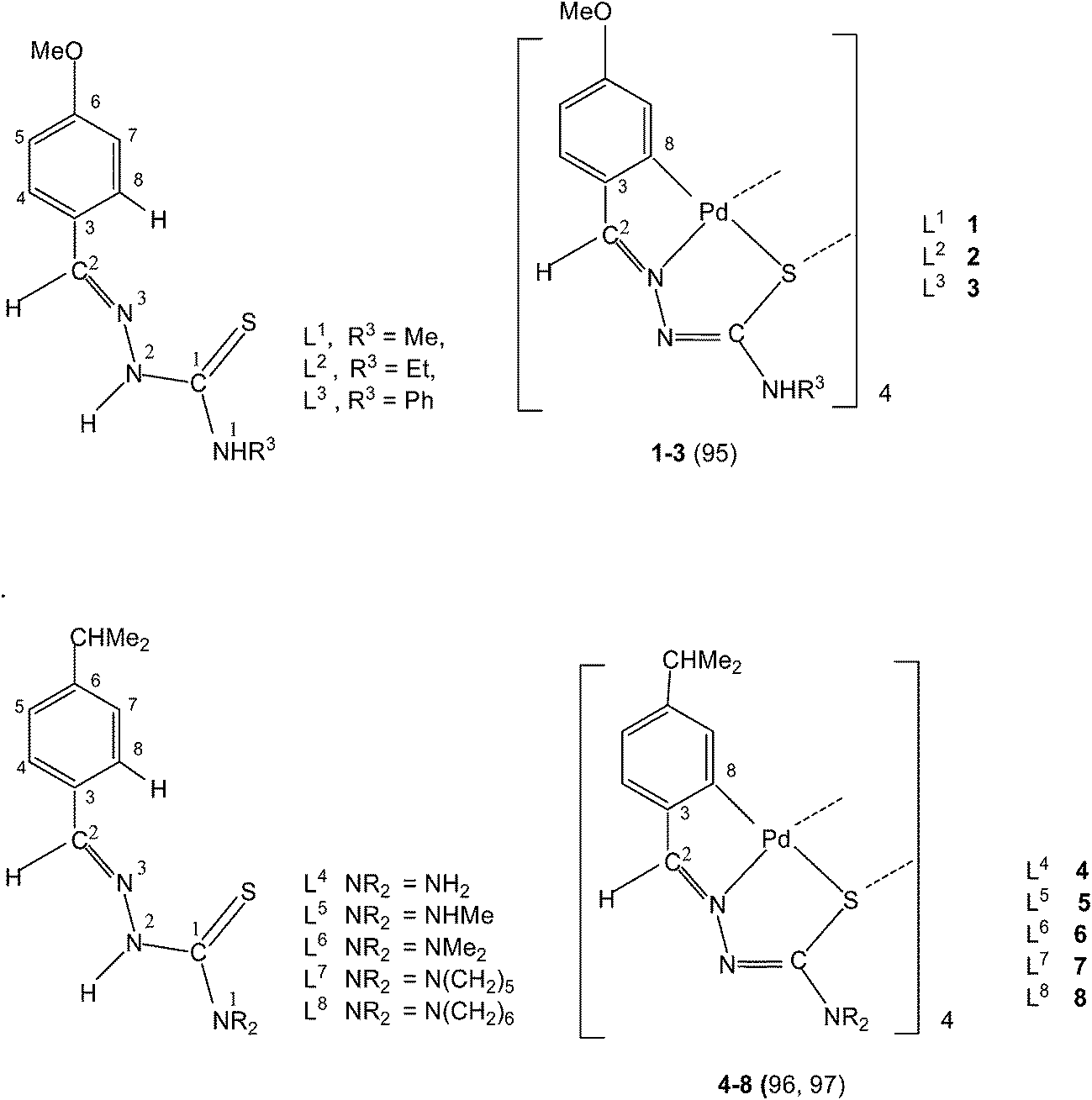
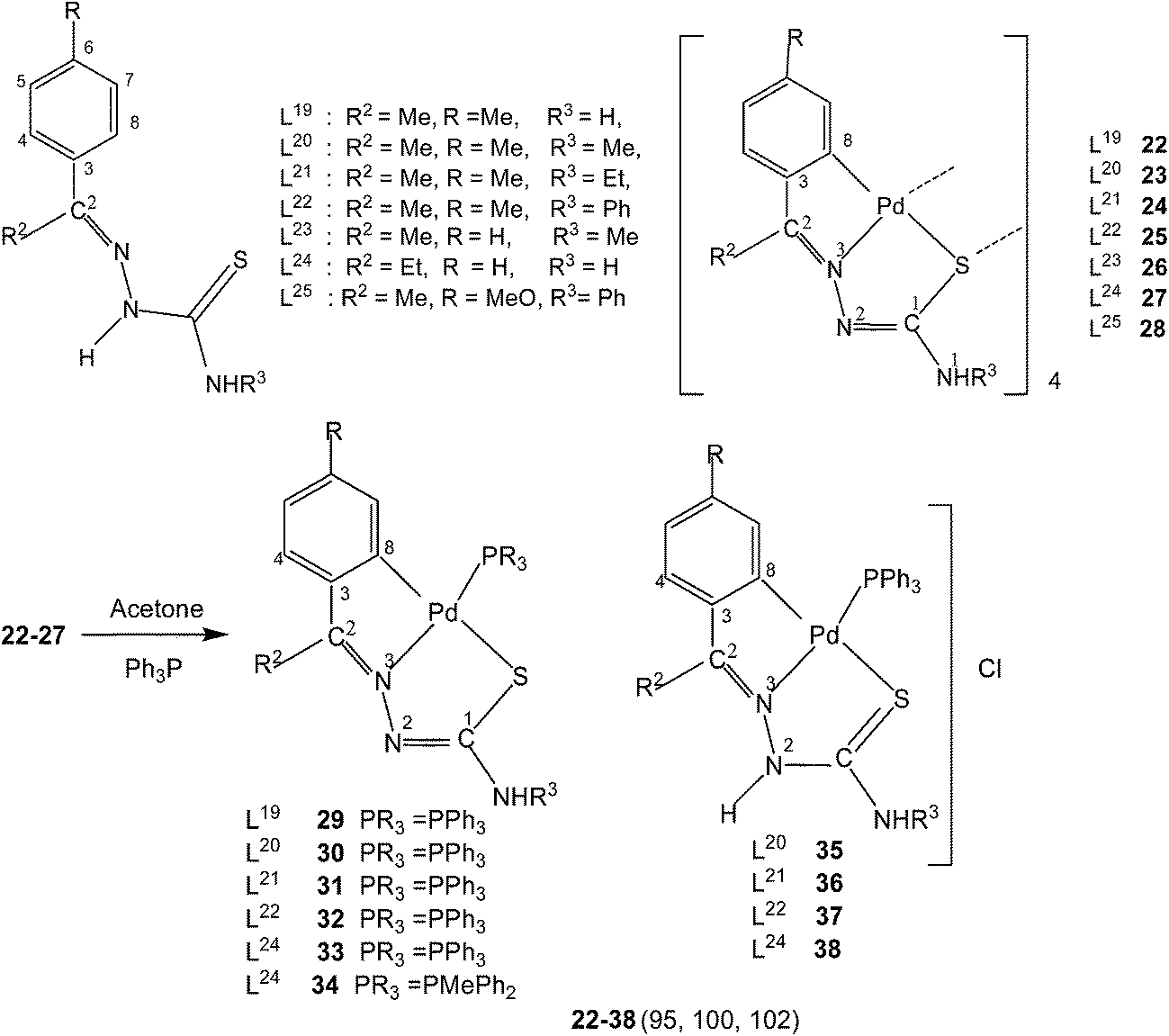
Variation of R1 groups at C2 carbon with R2 substituent as hydrogen. The presence of p-methoxyphenyl (R1) substituent at C2, R2 = H and N1R3R4 = N1HR3 gives rise to p-methoxyphenyl thiosemicarbazones with structure L1–3. Reactions of L1 and L2 with lithium tetrachloropalladate, in presence of sodium acetate in methanol solvent and that of L3 with potassium tetrachloropalladate in ethanol (molar ratio M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L: 1:2) yielded tetranuclear complexes, {Pd4(C,N,μ-S-L1–3-2H)4} (1–3).95 There is activation of C8–H hydrogen (ortho to C3) of p-methoxyphenyl moiety as well as that of imino–N2(H)–hydrogen and the thio-ligands coordinate to the metal as di-anions through terdentate C,N,S group. Interestingly, the shift in the position of methoxy group from para-position (C6) to meta-position (C5) when reacted with lithium tetrachloropalladate in methanol did not involve C–H activation and only N3,S-chelated cis-square planar complexes, cis-[Pd(N3,S-L)2] after deprotonation of N2–H hydrogen were obtained.95 Another series of thio-ligands, L4–8 with palladium(II) acetate in acetic acid also yielded tetranuclear complexes, {Pd(C,N,μ-S-L)}4 (L = L4–8) 4–8.96,97
:L: 1:2) yielded tetranuclear complexes, {Pd4(C,N,μ-S-L1–3-2H)4} (1–3).95 There is activation of C8–H hydrogen (ortho to C3) of p-methoxyphenyl moiety as well as that of imino–N2(H)–hydrogen and the thio-ligands coordinate to the metal as di-anions through terdentate C,N,S group. Interestingly, the shift in the position of methoxy group from para-position (C6) to meta-position (C5) when reacted with lithium tetrachloropalladate in methanol did not involve C–H activation and only N3,S-chelated cis-square planar complexes, cis-[Pd(N3,S-L)2] after deprotonation of N2–H hydrogen were obtained.95 Another series of thio-ligands, L4–8 with palladium(II) acetate in acetic acid also yielded tetranuclear complexes, {Pd(C,N,μ-S-L)}4 (L = L4–8) 4–8.96,97
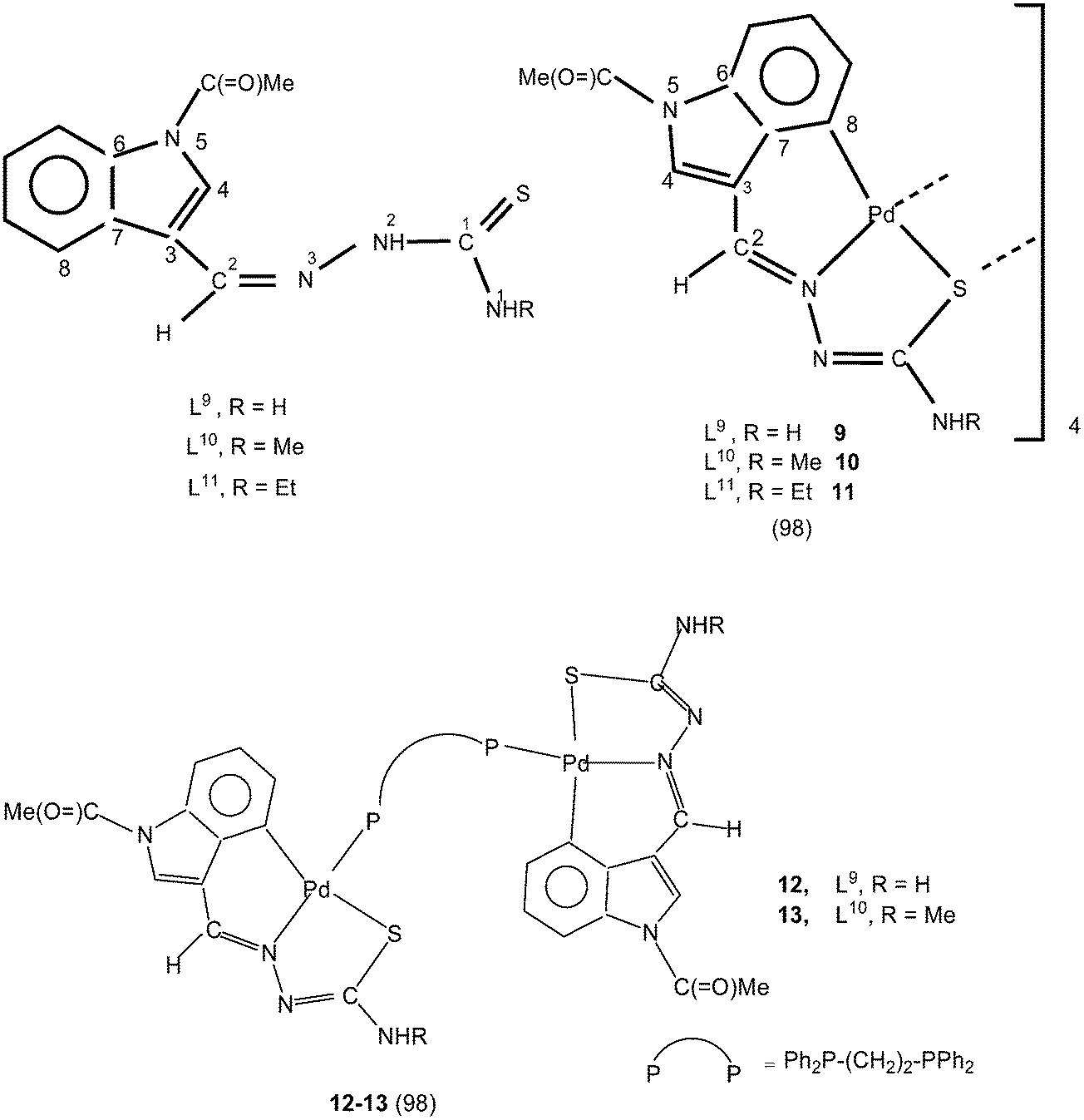
Equimolar reactions of N-acetyl-3-indolecarbaldehyde thiosemicarbazones (L9–11) with potassium tetrachloropalladate in water–ethanol have yielded cyclometallated tetranuclear complexes, {Pd4(C,N,μ-S-L9–11-2H)4} (9–11),98 which are stoichiometrically similar to 1–3. There is a six-membered metallacyclic ring unlike five-membered ring usually observed as for example in 1–8. Oligomers 6 (L9) and 10 (L10) suspended in acetone on reaction with a ditertiary phosphine, Ph2P–CH2–CH2–PPh2 (M:L ratio 1:2) formed P,P-bridged dinuclear complexes 12 (L9) and 13 (L10).98 The oligomerization occurred through coordinated sulfur in tetranuclear complexes 1–11.
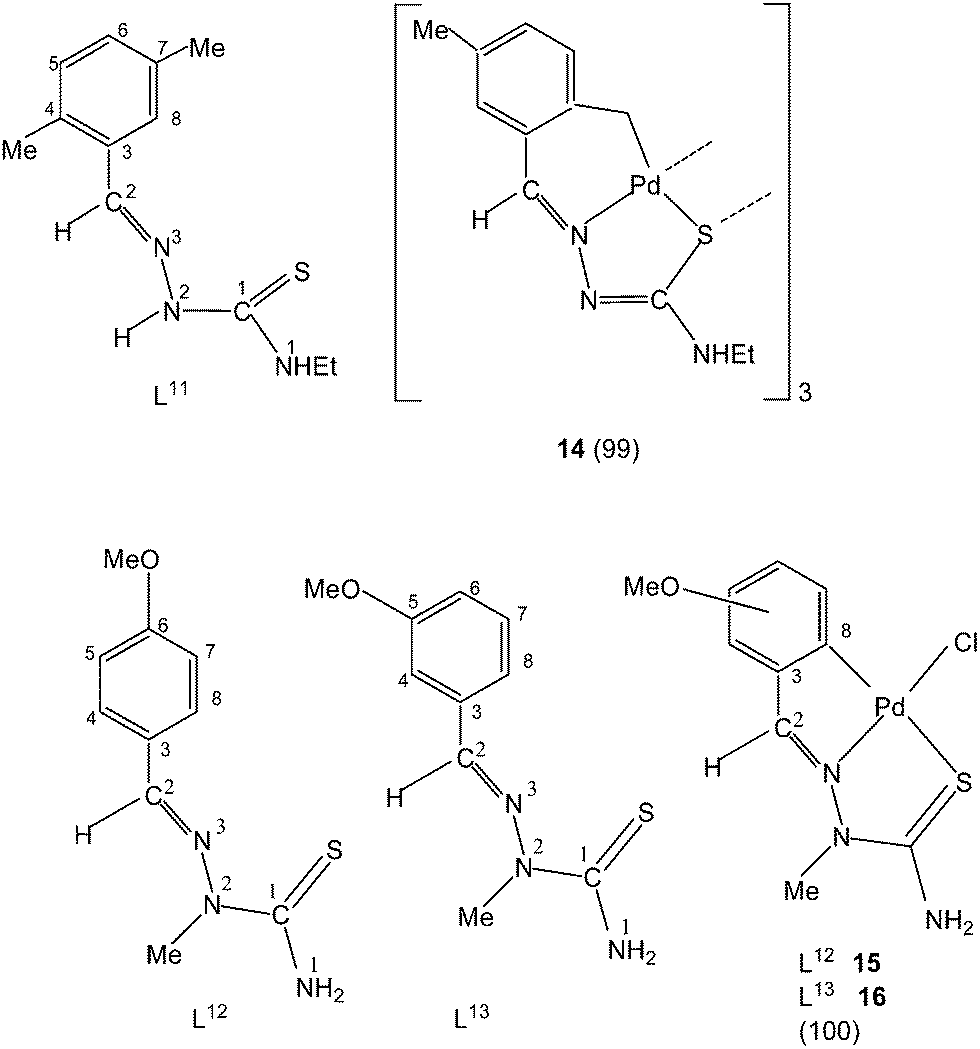
Thiosemicarbazone L11 with two methyl groups at ortho- and meta-positions of phenyl ring on reaction with palladium acetate in acetic acid yielded a trinuclear cyclometallated palladium(II) compound {Pd3(C,N,μ-S-L11-2H)3} 14.99 Each thiosemicarbazone ligand is tridentate with the metal bonded to the carbon atom from the o-methyl group (C4–Me), to the azomethine nitrogen and to the sulfur atom, which bridges to an adjacent palladium center. The central core Pd3S3 of the trinuclear complex confirms the presence of a non-planar six-membered metallacylic ring.99 Metallation of the aliphatic methyl group (sp3 hybrid carbon) is rare occurrence in metal-thiosemicarbazone chemistry.
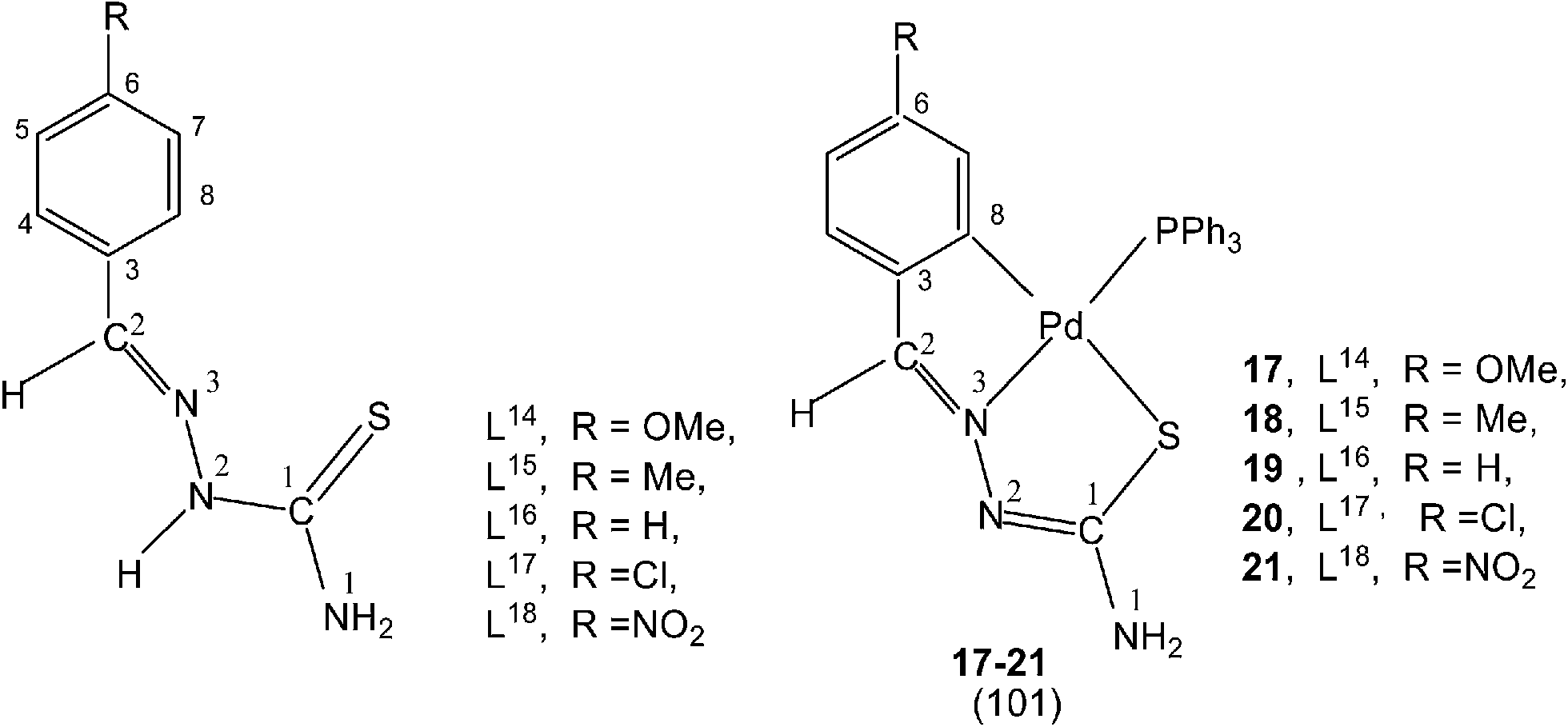
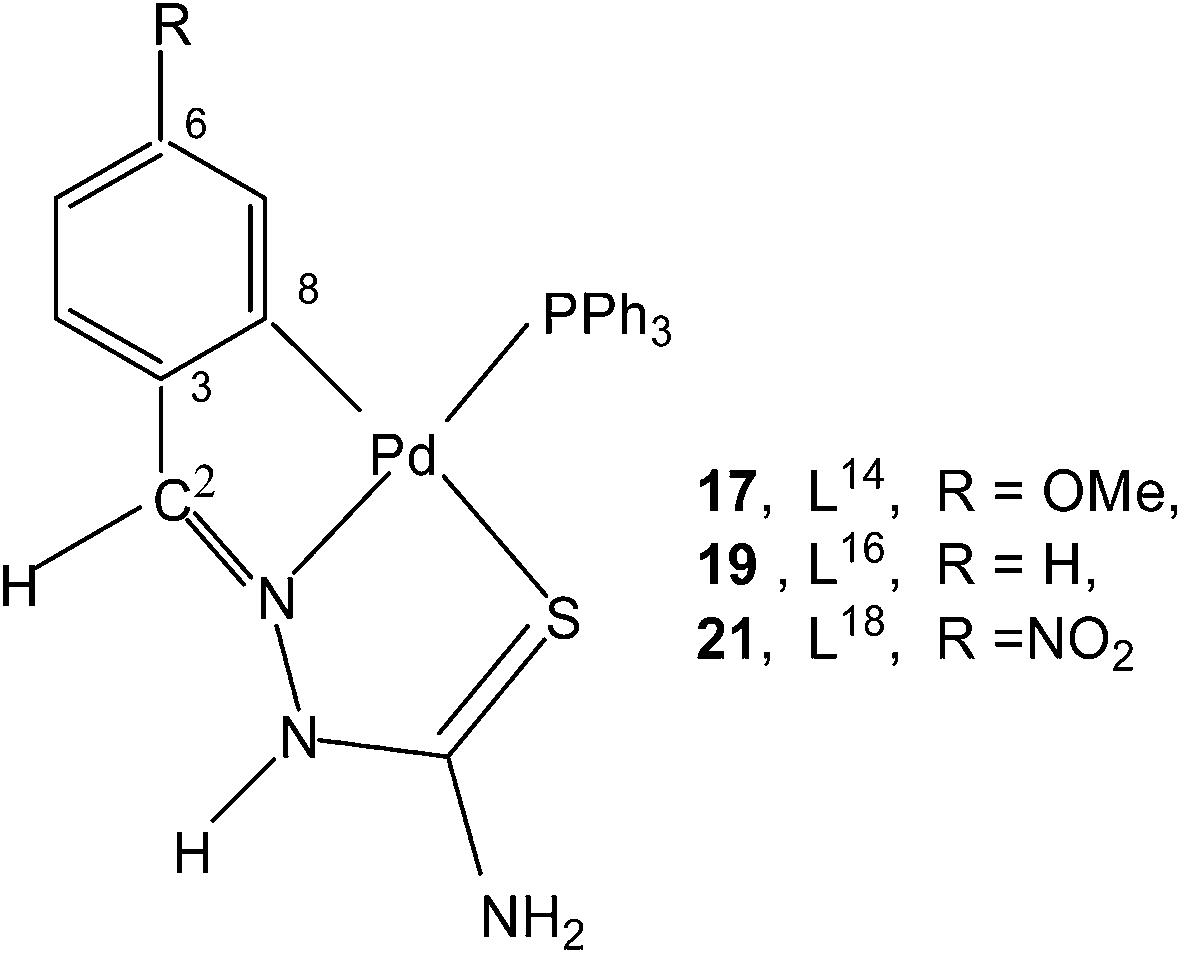
Reactions of thio-ligands L12–13 (methyl substituent at N2 atom instead of hydrogen) with potassium tetrachloropalladate in ethanol–water yielded mononuclear complexes, [Pd(C,N,S-L12–13-H)Cl] 15–16, with the ligand acting as mono-anion, C,N,S-terdentate.100 This behavior is in contrast to that shown by the ligands L1–3 which gave polynuclear complexes (1–3). Reactions of thio-ligands L14–18 with Na2PdCl4 in presence of 2-picolinic acid in presence of Et3N in hot ethanol formed two types of products, (a) cis-square planar complexes with Pd(N3,S-L)2 core and (b) brown polymeric complexes, [{Pd(CNS-L14–18-2H)}n].101 When these polymeric complexes were reacted with PPh3 in ethanol, mononuclear complexes 17–21 were obtained in which thio-ligands coordinate as terdentate C,N,S donors.101
Variation of R1 groups at C2 carbon with R2 substituent as other than hydrogen. In this section, palladium(II) chemistry of thio-ligands with different groups (R1, R2) at C2 and at N1 (R3, R4) atoms is described. Specifically, R2 group at C2 is other than hydrogen. Reactions of thiosemicarbazones L19–25 (with methyl/methoxy substituents in the phenyl ring and with Me/Et substituents at C2) with K2PdCl4 in water–ethanol {or Pd(OAc)2 in glacial acetic acid} gave tetranuclear complexes, {Pd4(C,N,μ-S-L-2H)4} (L = L19–24), 22–27; 100,102L = L25, 28 (ref. 95) in which thio-ligands acted as C,N,S-donor groups and oligomerization occurred through coordinated S donor atom. The oligomers 22–27 were suspended in acetone followed by the addition of a tertiary phosphine, Ph3P/PMePh2, which converted oligomers into mononuclear, [Pd(C,N,S-L)(PPh3)] complexes (29–34).100,102 The phosphine complexes 30–33 suspended in ethanol when treated with conc. HCl involved protonation of N2 nitrogen and the thio-ligands though remain C,N,S-terdentate but bear one negative charge and Cl− ion is the counter ion to balance the cationic species in ionic complexes, [Pd(C,N,S-L)(PPh3)]+Cl− (35–37,100 38 (ref. 102).
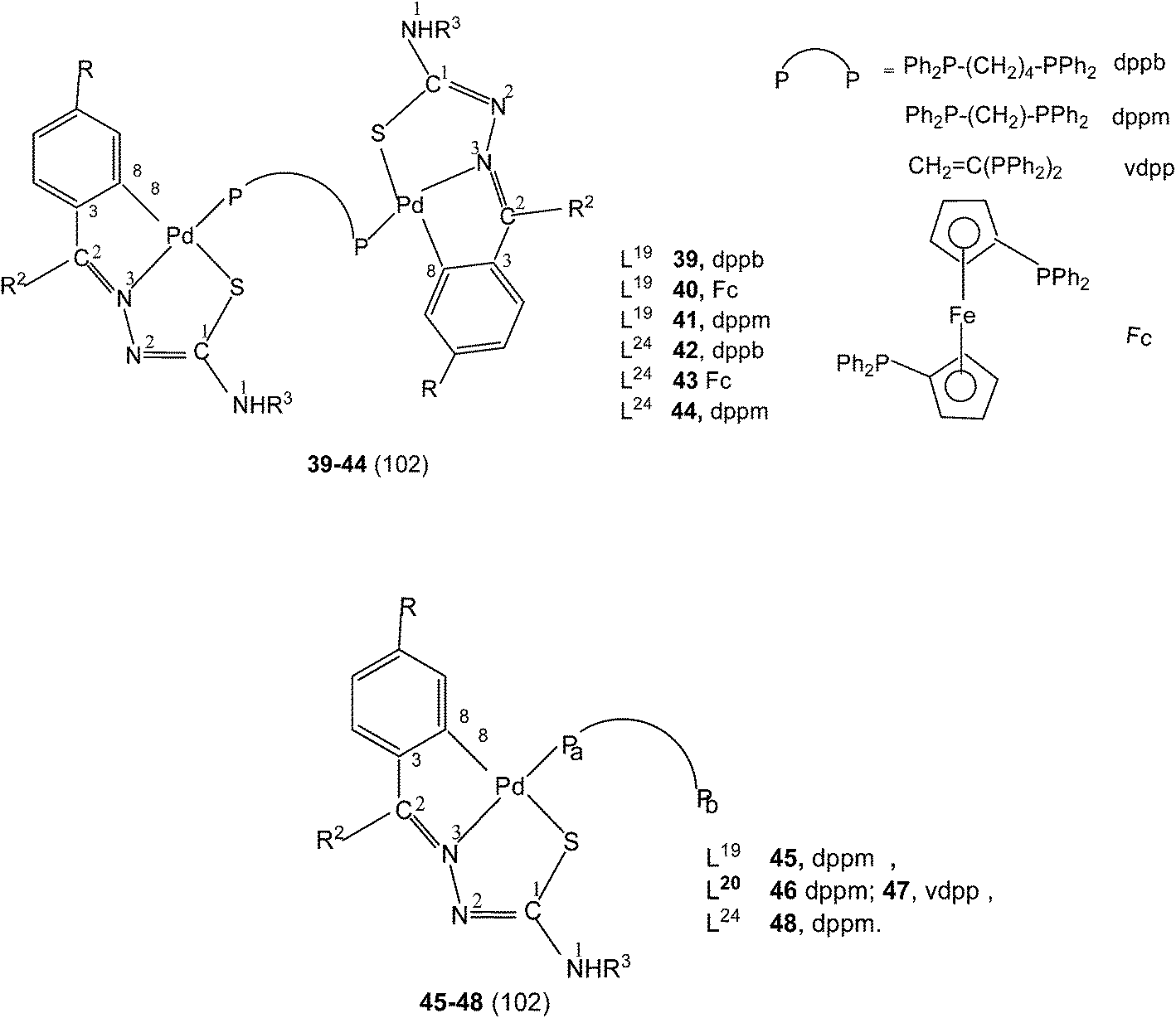
Further, tetranuclear complexes 22, 26 and 27 suspended in acetone with ditertiary phosphines (dppm, vdpp, dppb, and Fc) yielded dinuclear complexes (39–44) as well as mononuclear complexes with one pendant PPh2 group in each case (45–48).102 Reaction of tetranuclear complex 28 with Ph2P–CH2–PPh2, in acetone also formed P,P-bridged dimer, {Pd(C,N,S-L25)}2(μ-Ph2P–CH2–PPh2) and attempt to crystallize in chloroform led to the formation of a N3, S-chelated complex, cis-[Pd(N3,S-L25)2] with no metallation retained.95
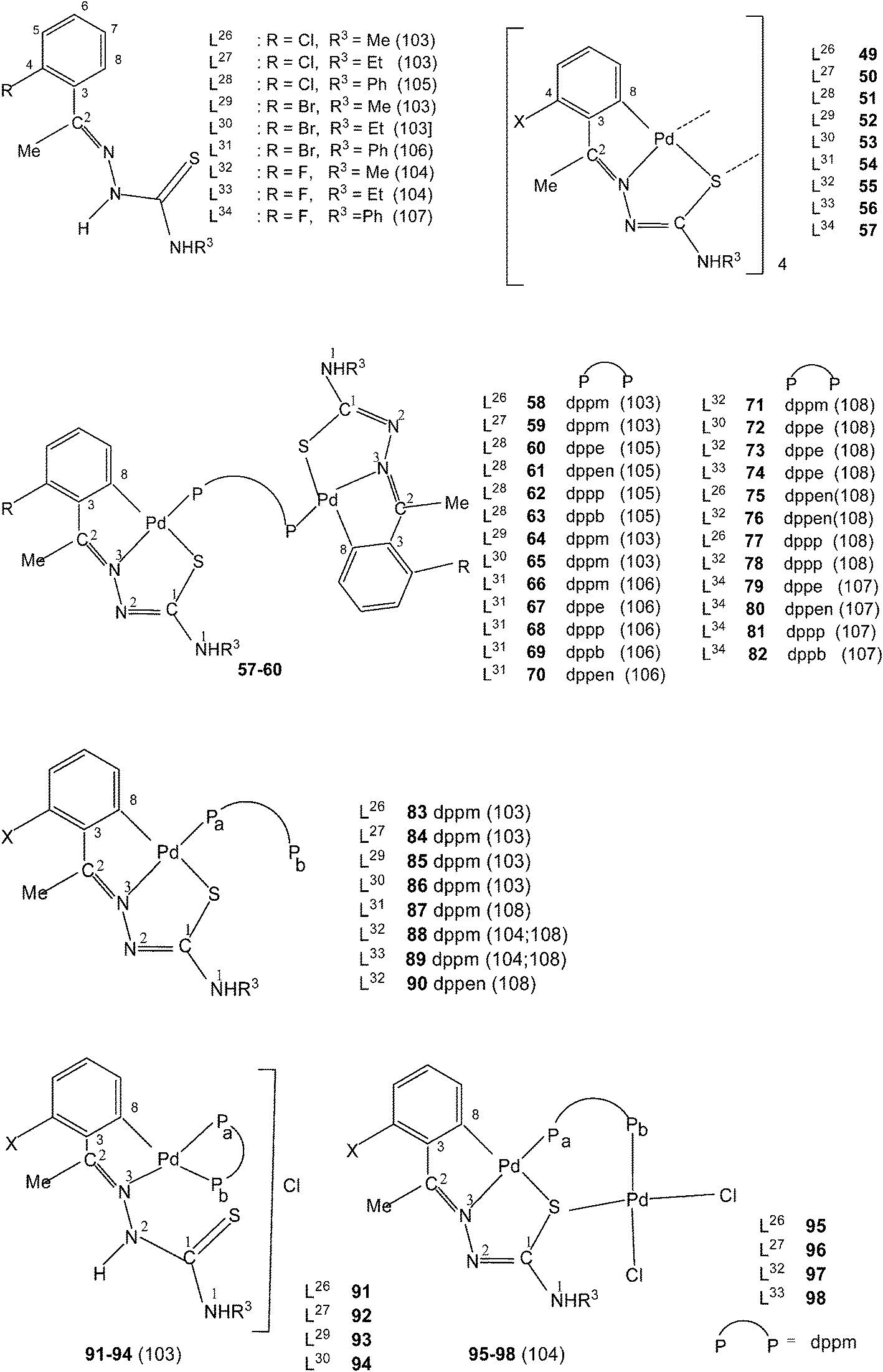
Ligands L26–34, having halogen substituents at 4-position of phenyl ring (R1) and with R2 substituents being Me, when reacted with Li2[PdCl4] in presence of sodium acetate in methanol solvent {or Pd(AcO)2} in acetic acid; K2PdCl4 in ethanol–water} also formed tetra-nuclear complexes,{Pd4(C,N,μ-S-L-2H)4}(L = L26–34; 49–57).103–108 Reactions of tetranuclear complexes 49–57 with diphosphines in acetone in 1:2 molar ratio formed phosphine bridged dinuclear complexes, {Pd(C,N,S-L)}2(μ-P,P-ligand) (P,P-ligand is a diphosphine) (58–82),103–108 and in 1:4 molar ratio in acetone yielded mononuclear complexes, 83–89 (dppm) and 90 (dppen). Complexes 83–86 when treated with aq. HCl in acetone formed ionic complexes, 91–94 with dppm as P,P-chelator. One –PPh2 group is pendant in each case. Complexes 83, 84, 88 and 89 were reacted with PdCl2(PhCN)2 in acetone which yielded hetero dinuclear complexes 95–98.104
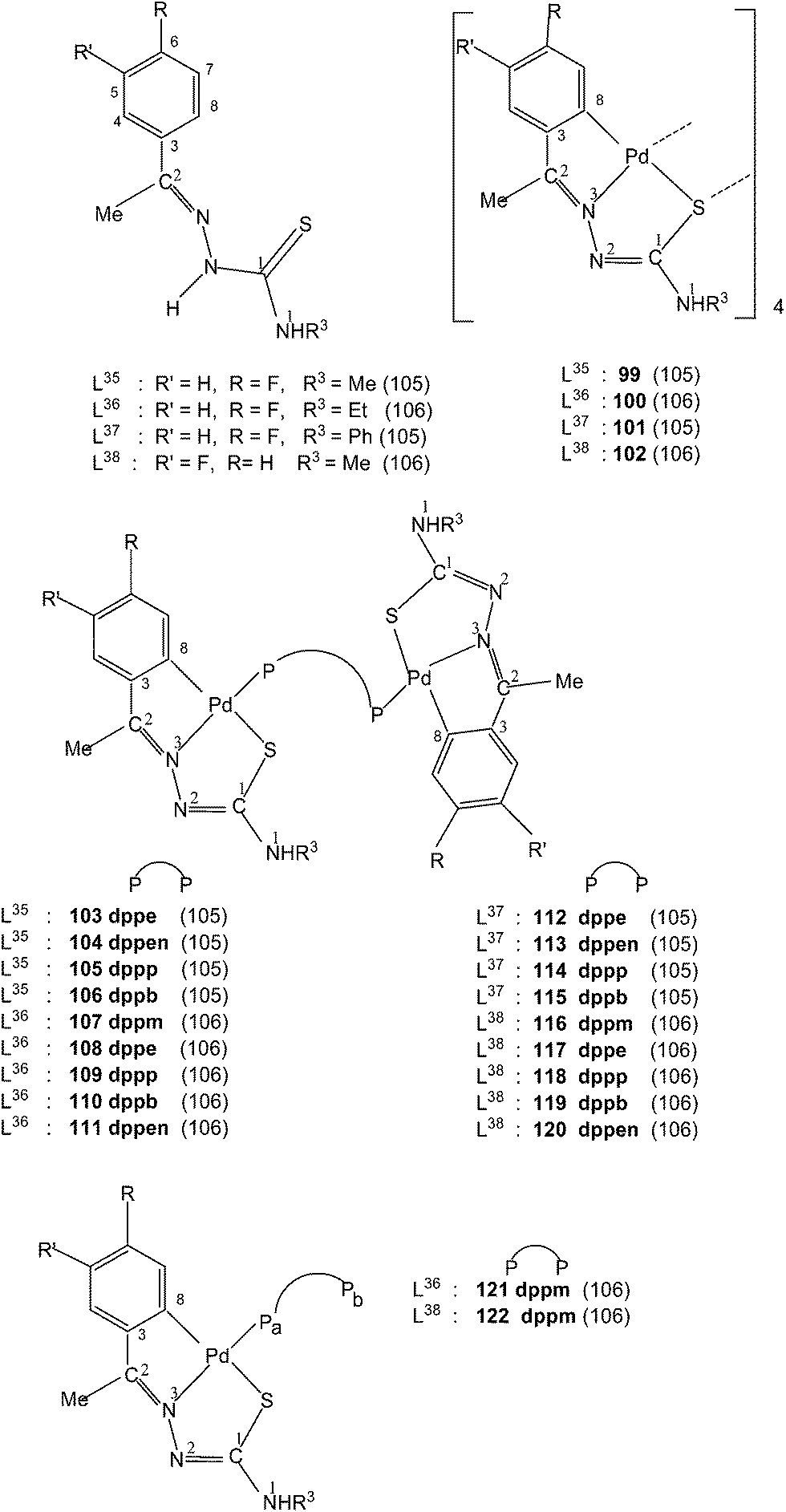
Ligands L35–38, having halogen substituents at 5- or 6-position of phenyl ring (R1) and with R2 substituents being Me, when reacted with Li2[PdCl4] in presence of sodium acetate in methanol solvent {or Pd(AcO)2} in acetic acid; K2PdCl4 in ethanol–water} also formed tetra-nuclear complexes,{Pd4(C,N,μ-S-L-2H)4} (L = L35–38; 99–102).105,106 Reactions of tetranuclear complexes 99–102 with diphosphines in acetone in 1:2 molar ratio formed P,P-bridged dinuclear complexes, {Pd(C,N,S-L)}2(μ-P,P-ligand) (P,P-ligand is a diphosphine) (103–120),105,106 and ligands, L36–38 with dppm in 1:4 molar ratio in acetone yielded mononuclear 121–122 complexes.106
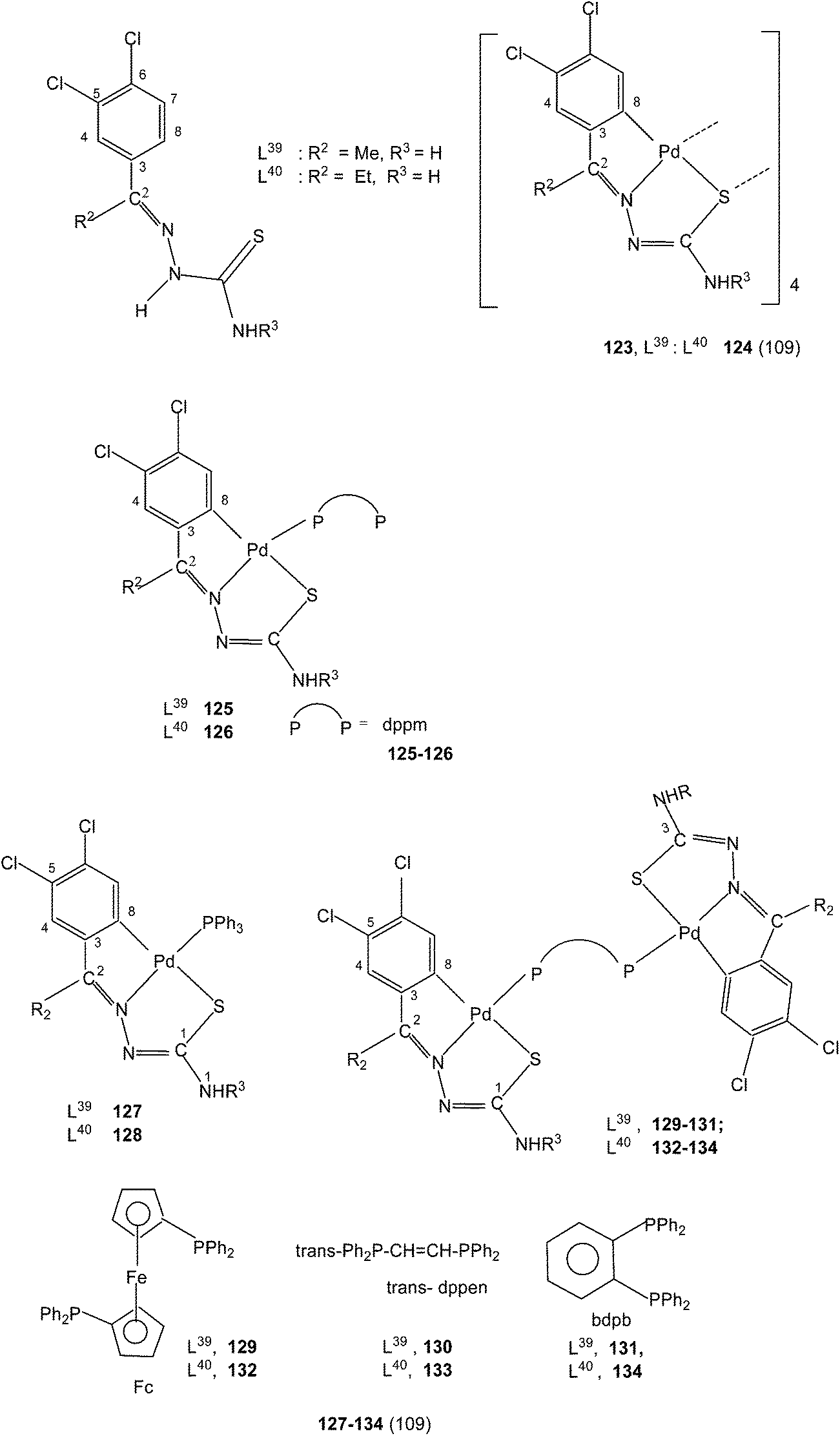
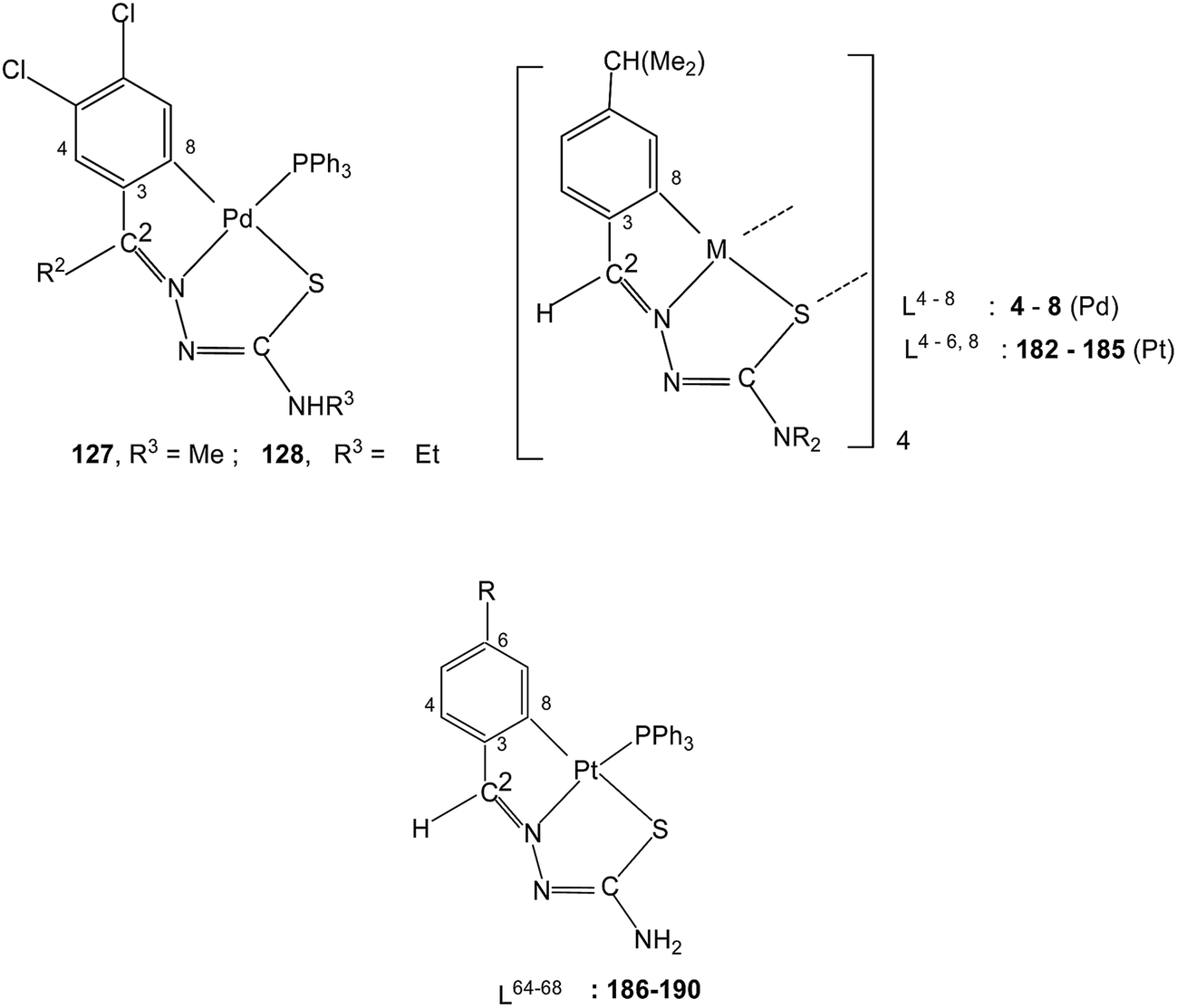
The presence of two halogens in the phenyl ring (R1) and with R2 being methyl/ethyl gave ligands L39–40 which on reaction with K2PdCl4 in ethanol–water again formed tetranuclear complexes 123 and 124 with C,N,S-terdentate thio-ligands.109 Tetranuclear complexes 123 and 124 with dppm in acetone formed mononuclear complexes 125 and 126 with one pendant –PPh2 group in each case. Compounds 123 and 124 with PPh3 in acetone yielded mononuclear complexes, 127 and 128 (ref. 109) Likewise 123 and 124 when reacted with ditertiary phosphines, namely, 1,1′-bis(diphenylphosphino)ferrocene (Fc), trans-bis(diphenylphosphino)ethylene (trans-dppen) and bis(diphenylphosphino)benzene (bdpb) formed dinuclear complexes, 129–134.109
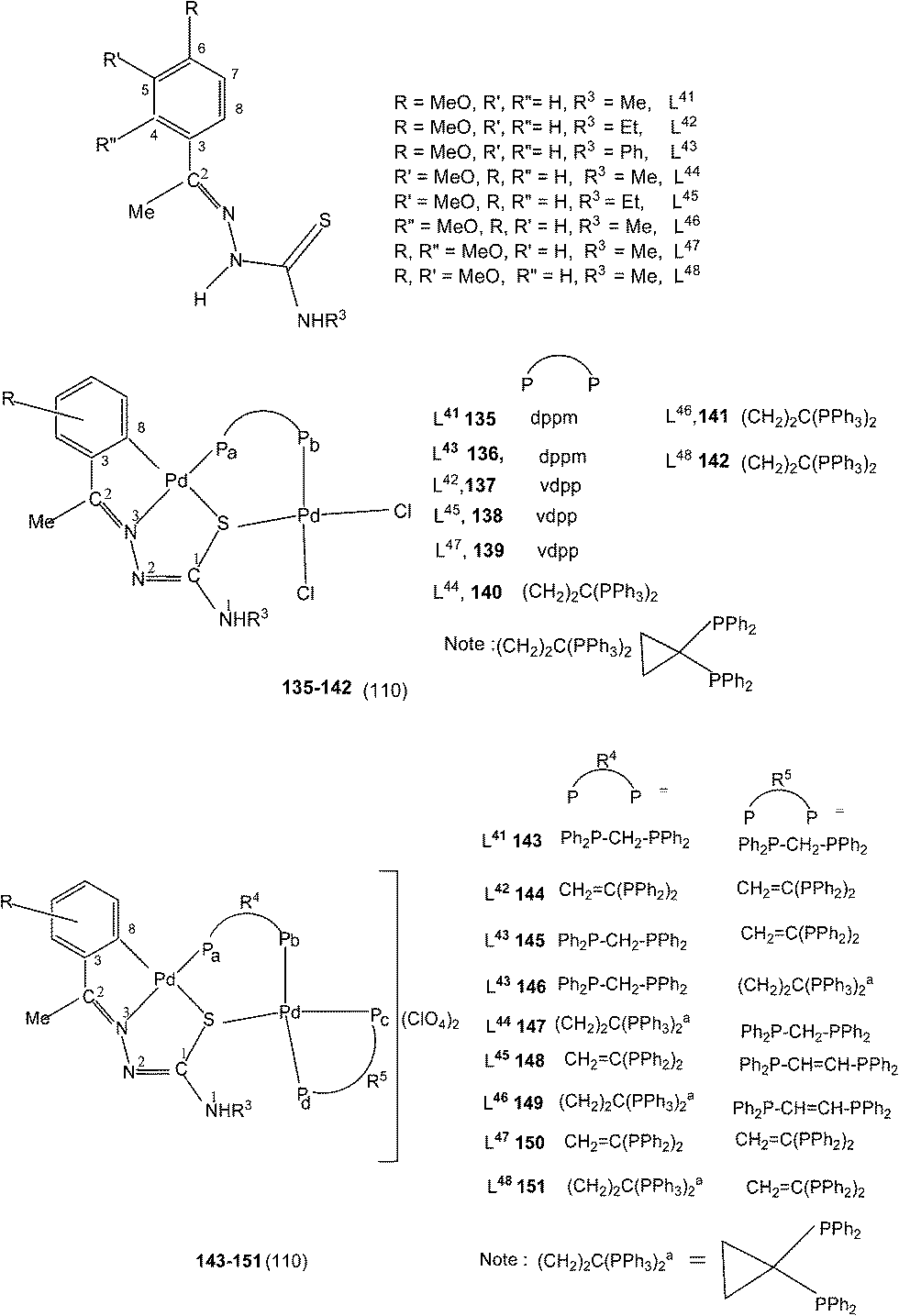
A series of ligands L41 to L48 having single or double methoxy substitution of phenyl ring also yielded dinuclear complexes, [Pd2Cl2(C,N,μ-S-L41–48) (μ-P,P-diphosphine)] 135 to 142,110 similar to complexes 95 to 98 as discussed earlier. Molecular structures of [Pd2Cl2(μ-C,N,S-L46) (μ-P,P-bdcp)] 141, [Pd2(μ-C,N,S-L41)(μ-P,P-dppm)(κ2-P,P-dppm)] 143 and [Pd2(μ-C,N,S-L44)(μ-P,P-bdcp)(κ2-P,P-dppm)] 147 {bdcp – bis(diphenylphosphino)cyclopropane},110 are determined. The dinuclear complexes with silver perchlorate in acetone gave ionic complexes 143 to 152.
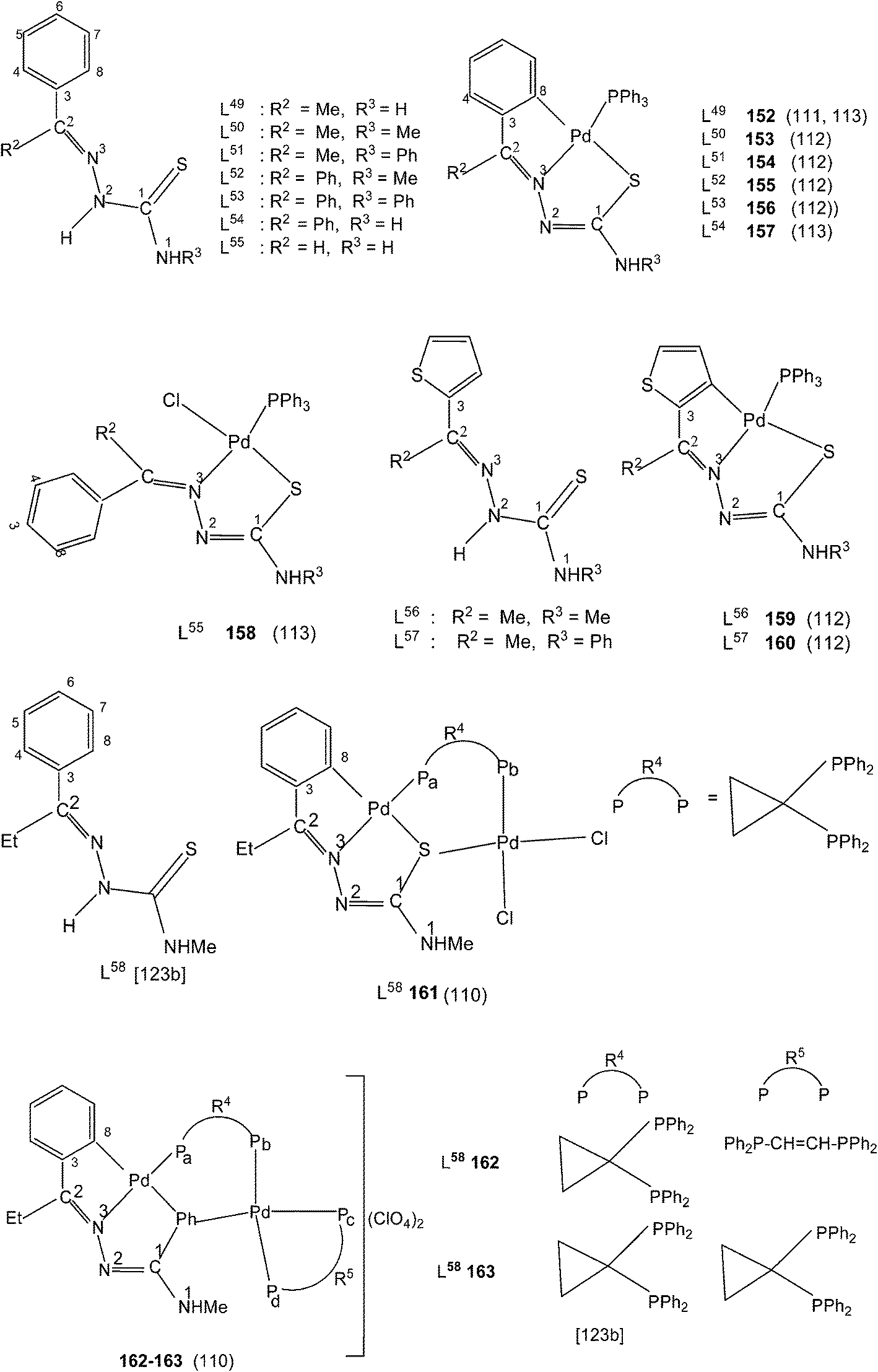
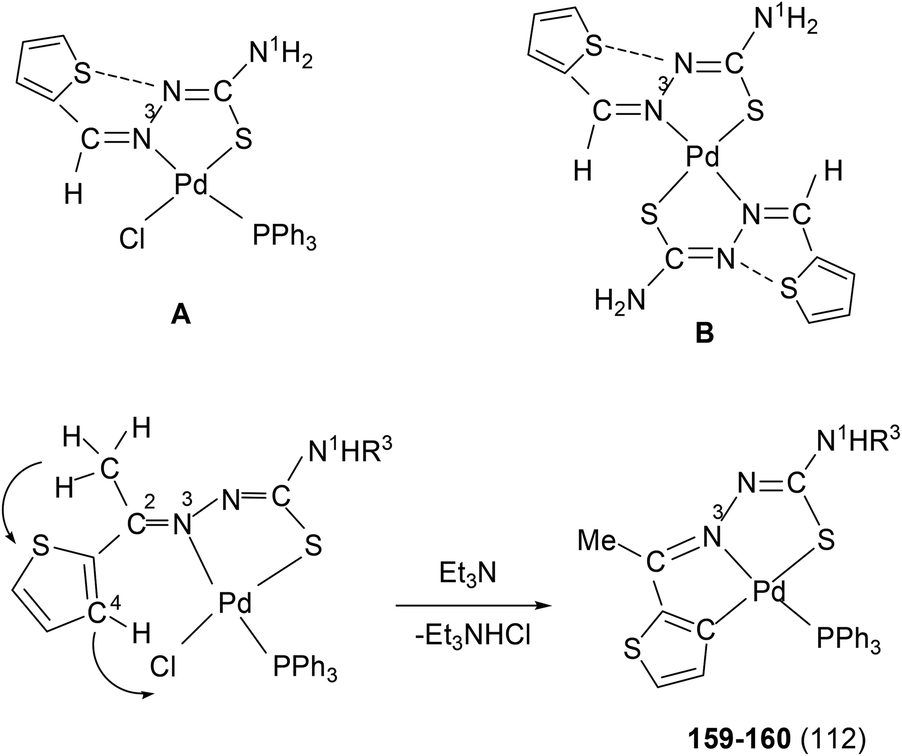
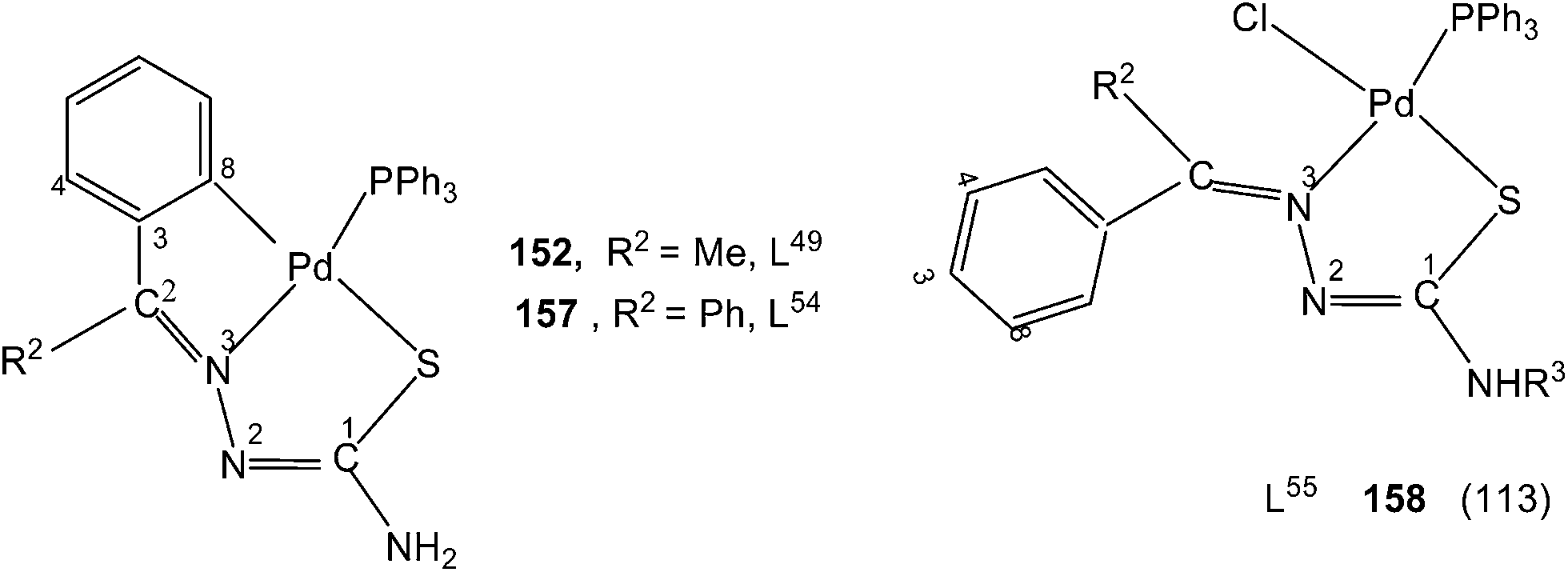
When there is no substituent in the phenyl ring (R1), but an aliphatic or aromatic substituent (R2) is present at C2 carbon and this gave rise to the thio-ligands L49–53 which on reaction with PdCl2(PPh3)2 in toluene (or acetonitrile) in presence of triethyl amine base has yielded only mononuclear square planar complexes, [Pd(C,N,S-L-2H)(PPh3)] (152–156)111,112 due to the presence of coordinated PPh3 which prevented oligomerization. Reactions were carried out in 1:2 (M:L) molar ratio (152),111 or 1:1 molar ratio (153–156),112 the latter ratio appears to work satisfactorily. The ortho-metallated complexes 152, 157 as well N3,S-chelated 158 were obtained from the reaction of PdCl2(PPh3)2 with the ligands L49,54,55 in ethanol in the presence of the base Et3N.111,113 When R1 group was thiopheneyl and with R2 = Me, using the same reaction method metallation similar to complexes 153–156 occurred and formed complexes 159–160 (L56–57).112 Pyrrole group (R2) at C2 did not form Pd–C bonds, rather pyrrole –NH moiety showed its deprotonation, and formed N,N,S-coordination in [Pd(N,N,S-L56–57) (PPh3)]114 unlike C,N,S-coordination shown in 159–160 (L56–57). The ligand L58 with a ethyl substituent at C2 has also formed dimeric complexes 161–163,110 similar to complexes 135 or 143.
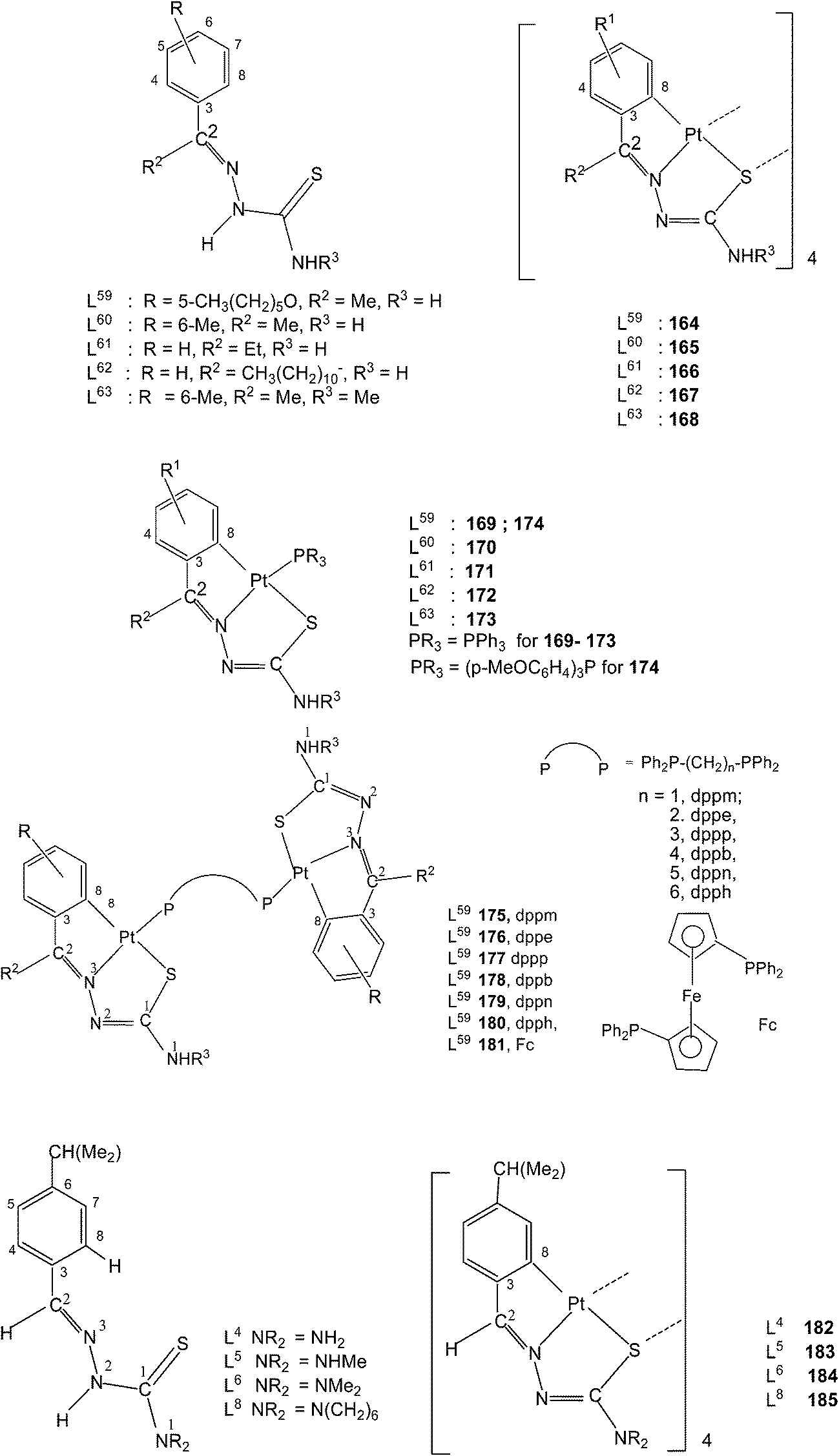
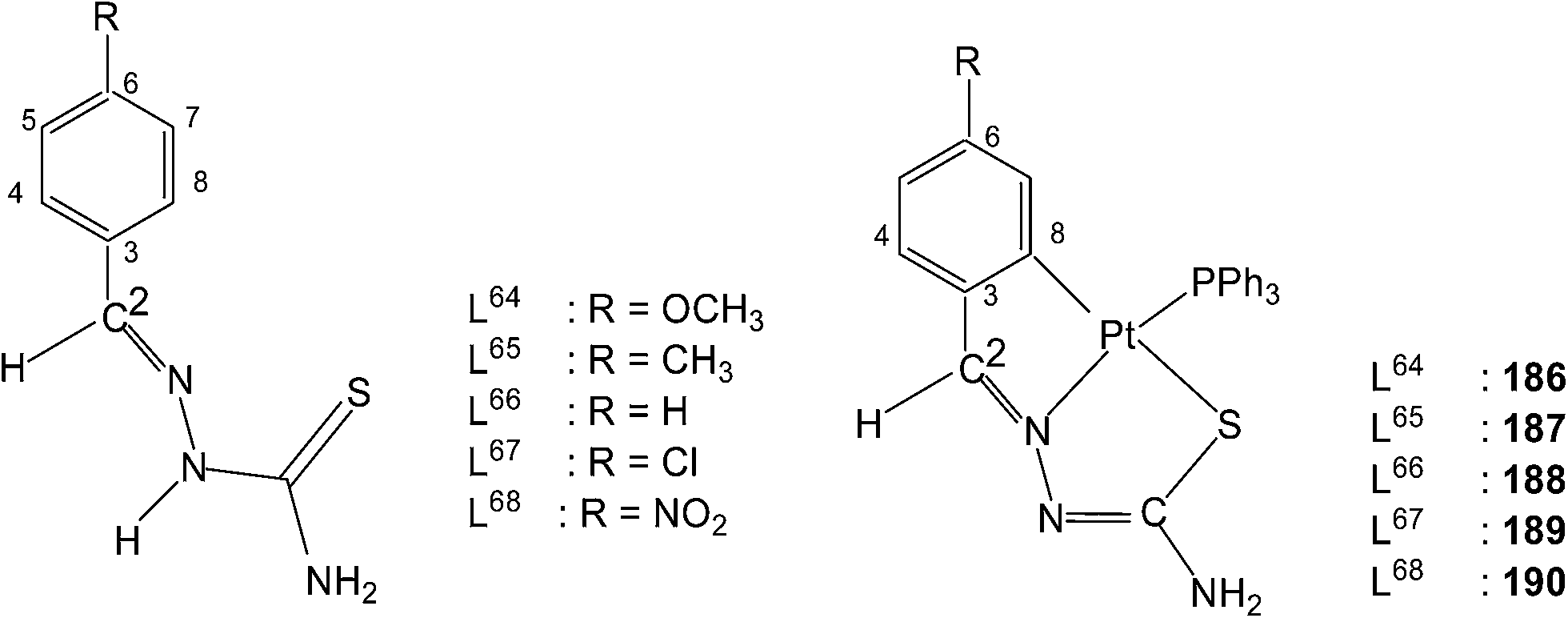
Platinum. Platinum(II) has also shown cyclometallation of coordinated thiosemicarbazones in a manner similar to that displayed by palladium(II), though number of reactions reported in comparison are much less. In a typical example, reaction of K2PtCl4 with the thio-ligand, L59 with R2 as Me substituent at C2 in water–ethanol mixture gave an orange tetranuclear complex, [Pt(C,N,S-L59-2H)]4 164.115 Similarly, with other ligands, L59–63 having different R2 substituents at C2, tetra-nuclear complexes, 165–168 were obtained.115 Treatment of oligomers 164–168 with PPh3 in 1:4 molar ratio in acetone followed by crystallization from acetone-n-hexane gave mononuclear complexes, [Pt(C,N,S-L-2H)(PPh3)] (169–173). With tri(p-methoxyphenyl)-phosphine, it formed a similar monomer, 170. Dinuclear complexes 174–181 were formed from tetranuclear complex 164 using various ditertiary phosphines. A series of thio-ligands, L4–6,8 with [Pt(μ-Cl)(η-C4H7)]2 in acetone or K2PtCl4 in methanol yielded tetranuclear complexes, {Pt(C,N,μ-S-L)}4 (L = L4–6,8) 182–185.96,97
Reactions of each of thio-ligands L64–68 with PtCl2(PPh3)2 in ethanol in presence of triethyl amine (1:1 molar ratio) for a period of 6 h gave cyclometallated complexes, [Pt(C,N,S-L-2H)(PPh3)] (186–190) in which the thio-ligands as dianions are C,N,S-donors.116 Interestingly, when these ligands were reacted with K2PtCl4 in methanol in presence of triethyl amine (2:1: L:M molar ratio) for 24 h, no metallation occurred, rather cis-square planar N3,S-chelated complexes cis-[Pt(N3,S-L)2] were obtained. Here the thio-ligands are coordinating as mono anions (losing N2–H proton).116
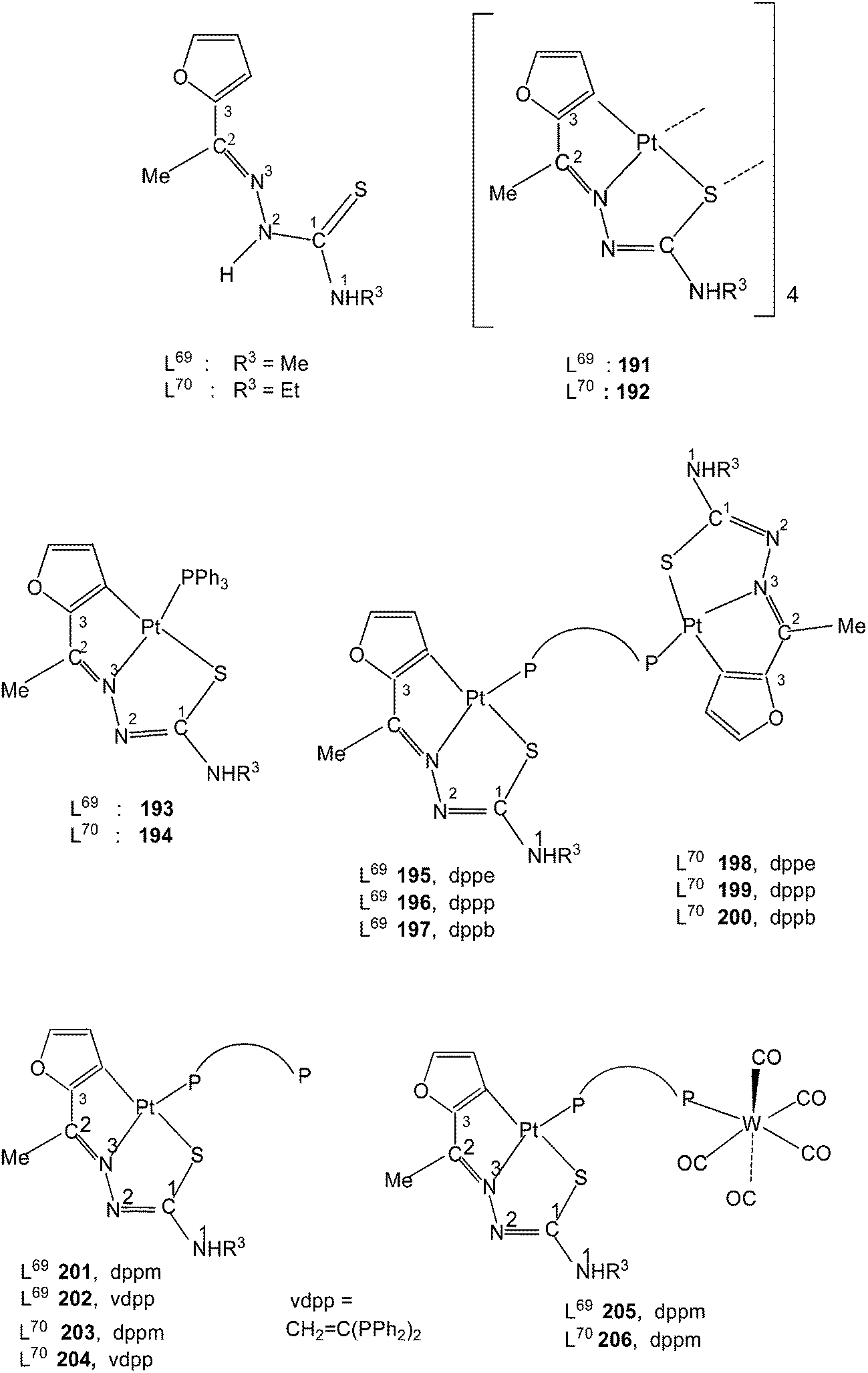
The thio-ligands L69–70 were refluxed with [cis-PtMe2(cod)] (1:1 molar ratio) in n-octane under nitrogen atmosphere which gave orange tetra nuclear complexes, [Pt(C,N,S-L-2H)]4 191 and 192.117 To a suspension of 191/192 in chloroform was added PPh3 (1:4 molar ratio) followed by heating for a short time yielded mononuclear complexes 193 and 194. Similar reactions with ditertiary phosphines having longer P,P bite, namely, dppe, dppp and dppb formed dinuclear complexes, 195–200. With dppm and vdpp, mononuclear complexes, 201–204, with one pendant –PPh2 group were obtained. To a suspension of 191/192 in chloroform was added [W(CO)5(Ph2P–CH2–PPh2)] (1:4 molar ratio) followed by heating for a short time which yielded hetero dinuclear complexes, 205 and 206. Treatment of 201 and 203 with [W(CO)5(THF)] also gave same products 205 and 206.
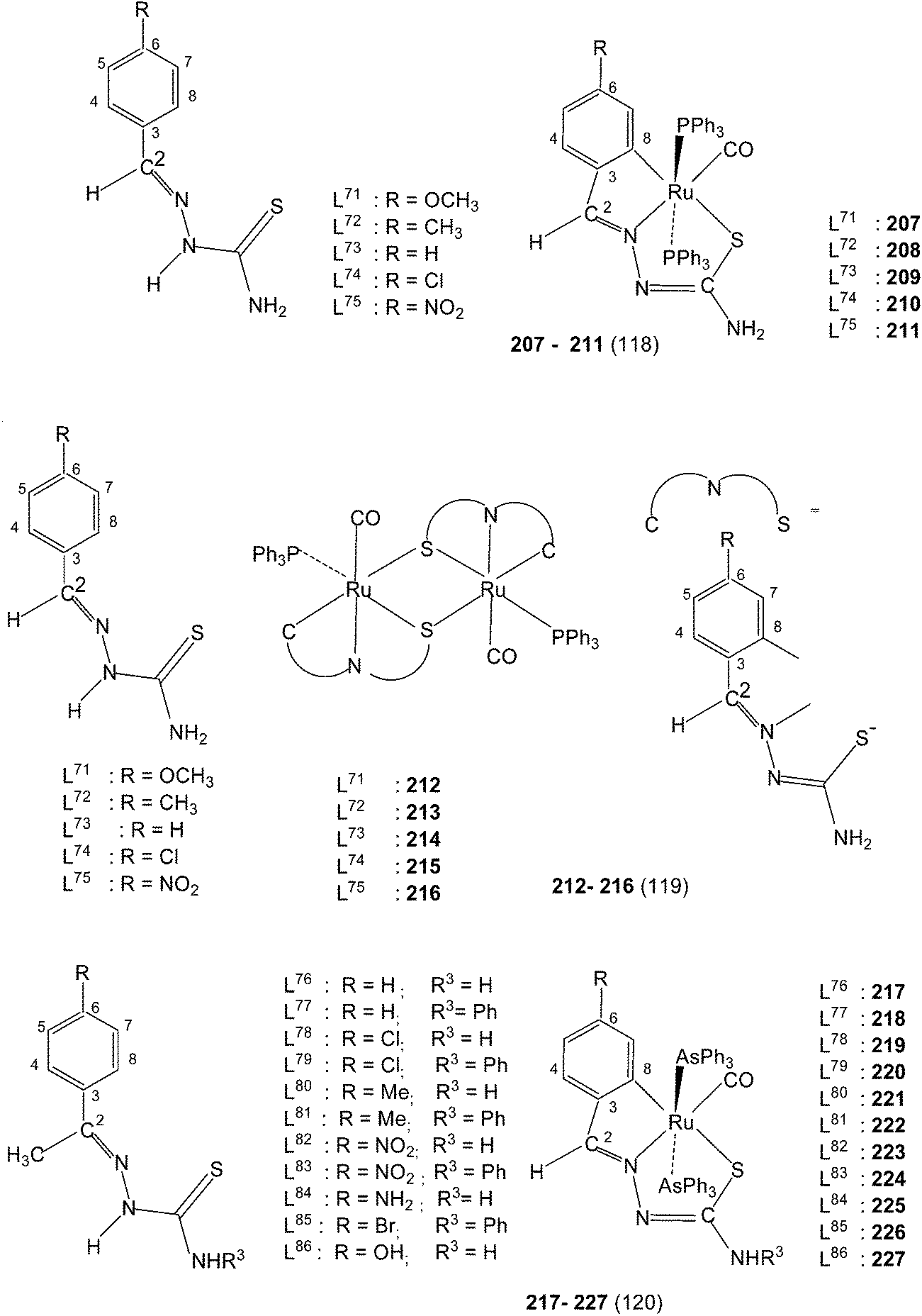
Ruthenium. Ruthenium is another metal which has shown cyclometallation. Reactions of thio-ligands L71–75 with [Ru(PPh3)3(CO)HCl] in refluxing methanol in presence of triethylamine afforded two types of complexes which were separated by thin layer chromatography.118 One class of complexes were having C,N,S-donors, [Ru(C,N3,S-L71–75-2H) (PPh3) (CO)] 207–211 and second class of complexes have N2,S-chelation with no cyclometallation, namely, [Ru(N2,S-L-H)(PPh3)2(H)CO]. Both PPh3 ligands are trans in both category of complexes. Hydrogen is trans to sulfur in second category of complexes and CO is trans to N2/N3 atoms. Reactions of thio-ligands L71–75 with [Ru(PPh3)2(CO)2Cl2] in refluxing toluene yielded dinuclear complexes [Ru2 (C,N,S-L)2(PPh3)2(CO)2] (212–216).119 When these reactions were carried out in refluxing ethanol, only complexes [Ru(N2,S-L-H)(PPh3)2(H)CO] were formed in good yield with no cyclometallation. Here the source of hydride bonded to RuII is believed to be ethanol.119 Reactions of [RuHCl(CO)(AsPh3)3] with the thio-ligands L76–86 in presence of LiBr/NaOAc in methanol yielded a series of RuII complexes [Ru(C,N,S-L76–86)(CO(ASPh3)2)] 217–227.120
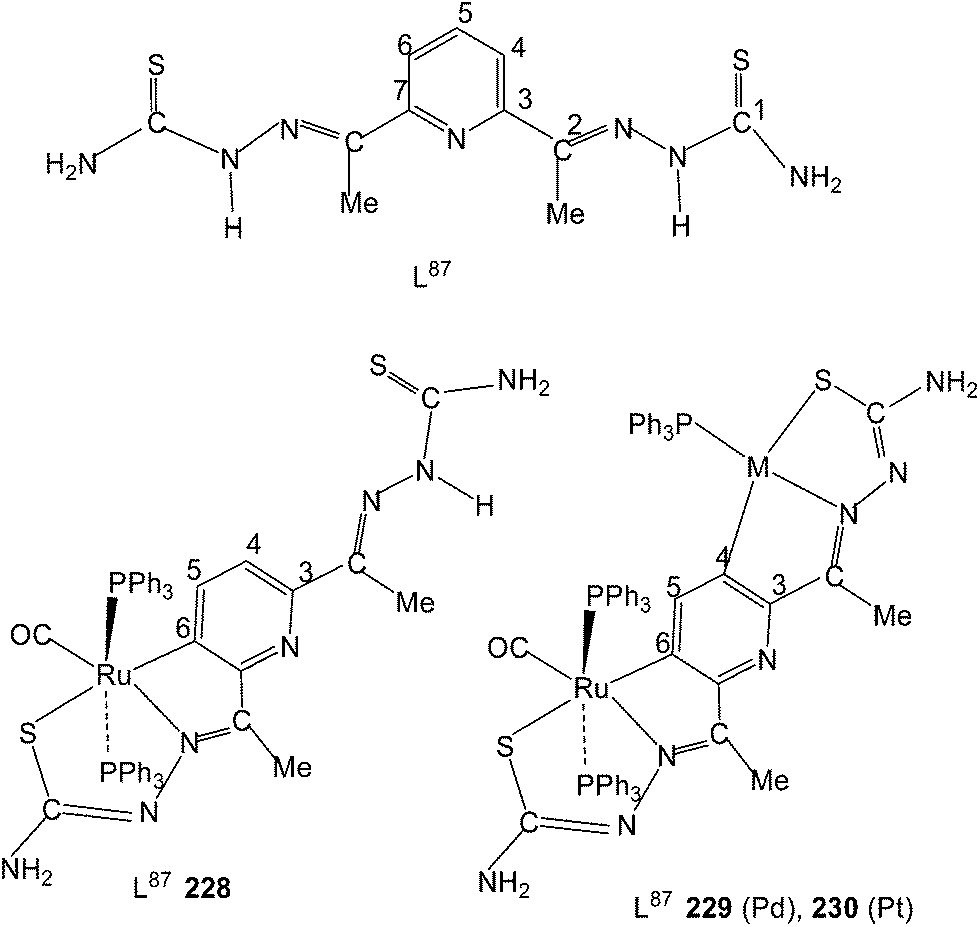
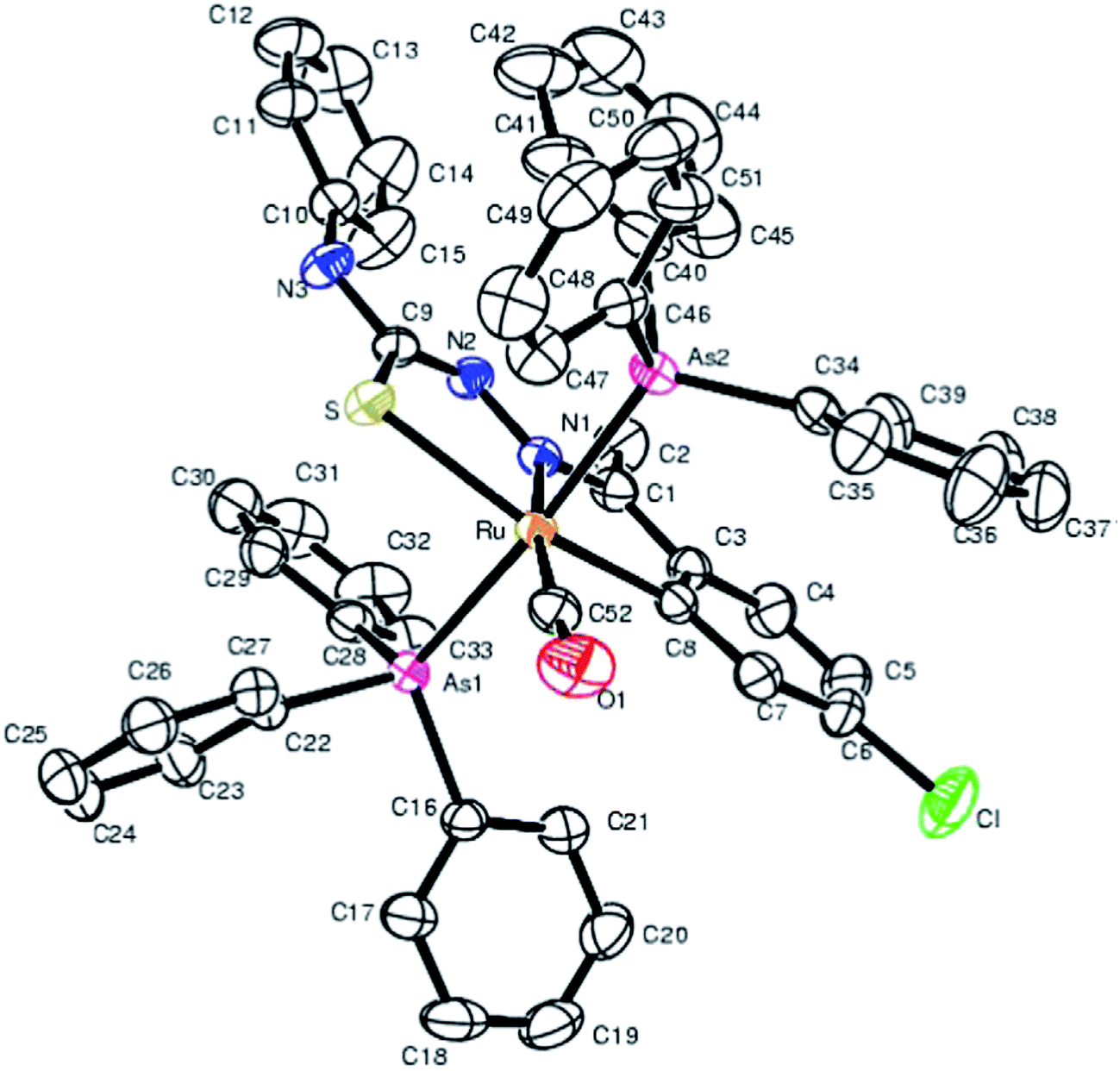
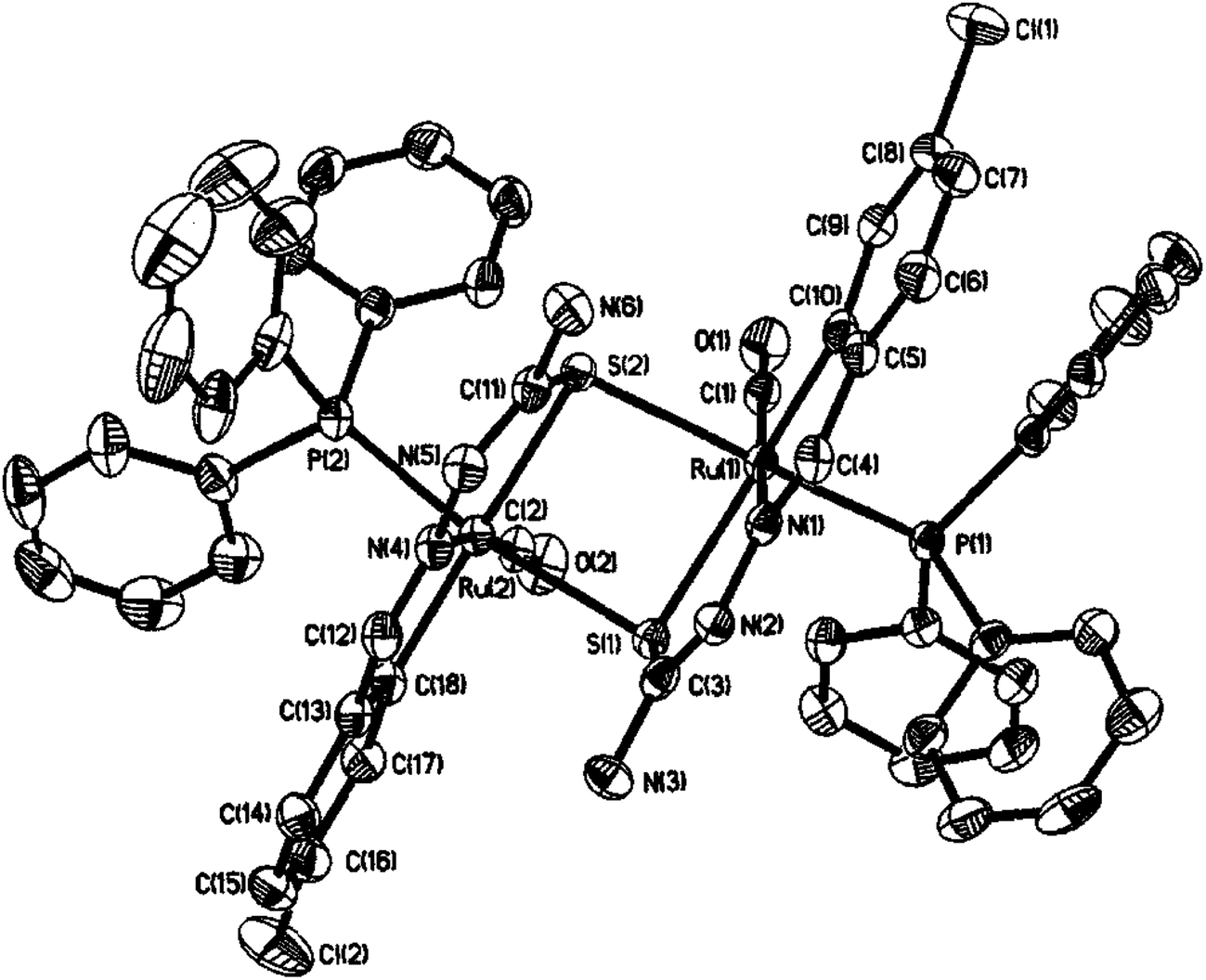
Equimolar reaction of [Ru(PPh3)2(CO)2Cl2] with 2,6-diacetylpyridine thiosemicarbazone (L87) {only example of a bis(thiosemicarbazone) involved in metallation} in refluxing ethanol followed by purification of orange solid using thin layer chromatography gave complex, [Ru(CO)(PPh3)2(C,N,S-L87-2H)] 228 in which pyridine ring is metallated and the ligand is dianion coordinating through C,N and S donor atoms.121 Further, reaction of organo ruthenium complex 228 with M(PPh3)2Cl2 (M = Pd, Pt) in ethanol followed by purification with TLC gave hetero dinuclear complexes, [RuM{(CNS)2-L87-4H}(CO)(PPh3)3] 229 (M = Pd), 230 (M = Pt) in which the second arm of the thio-ligands metallates Pd/Pt. Thus both 4 and 6 positions of the pyridine ring are metallated. Molecular structures of 228 and 230 are confirmed by X-ray crystallography and that of complex 229 with the help of DFT calculations.
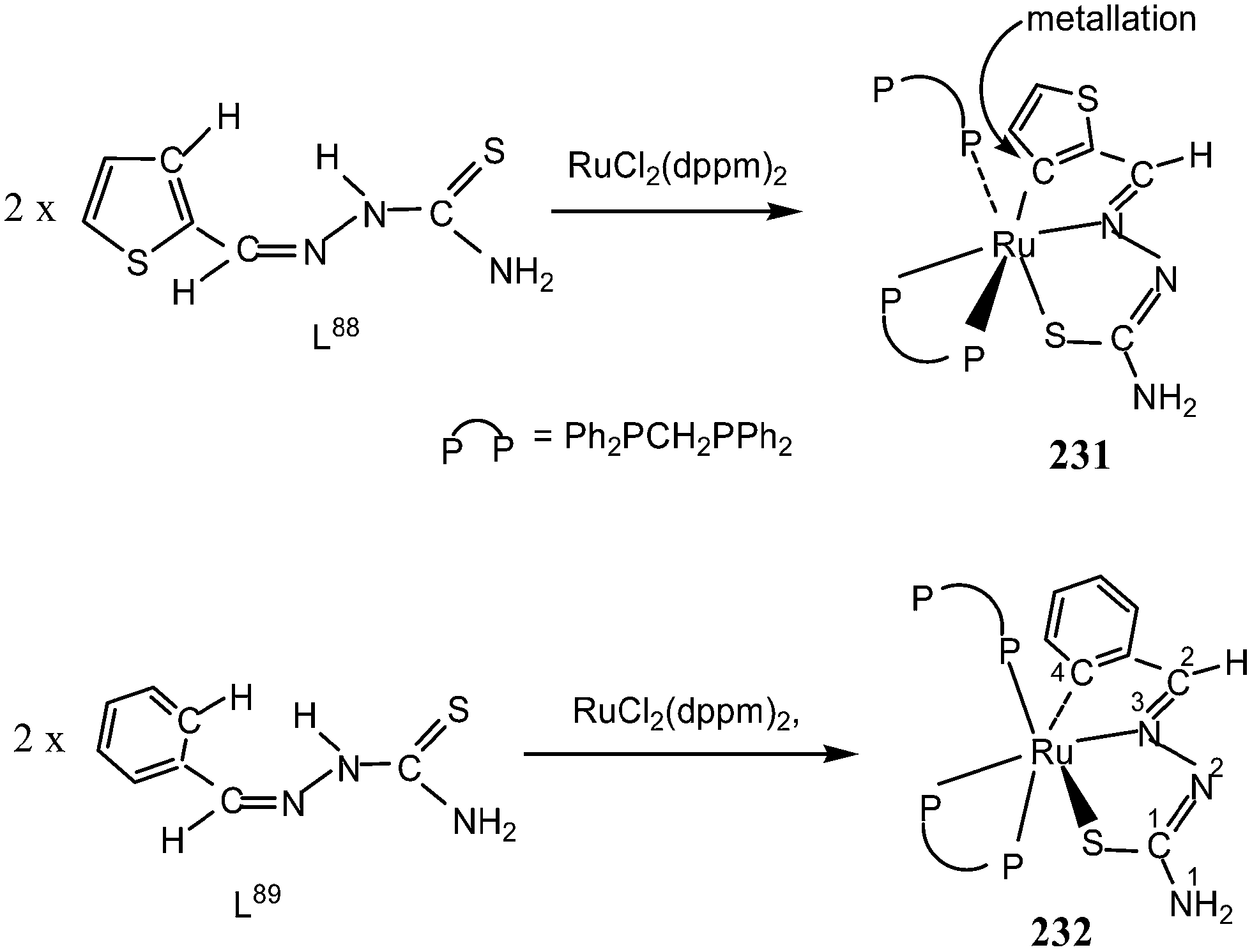
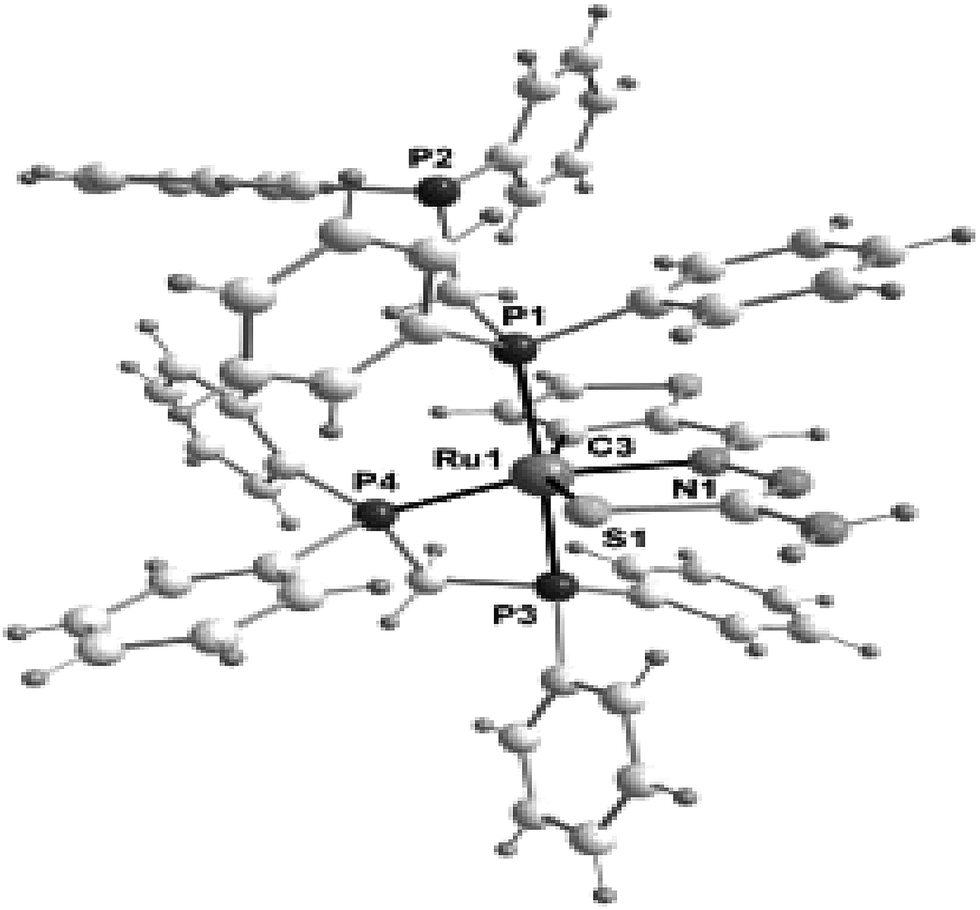
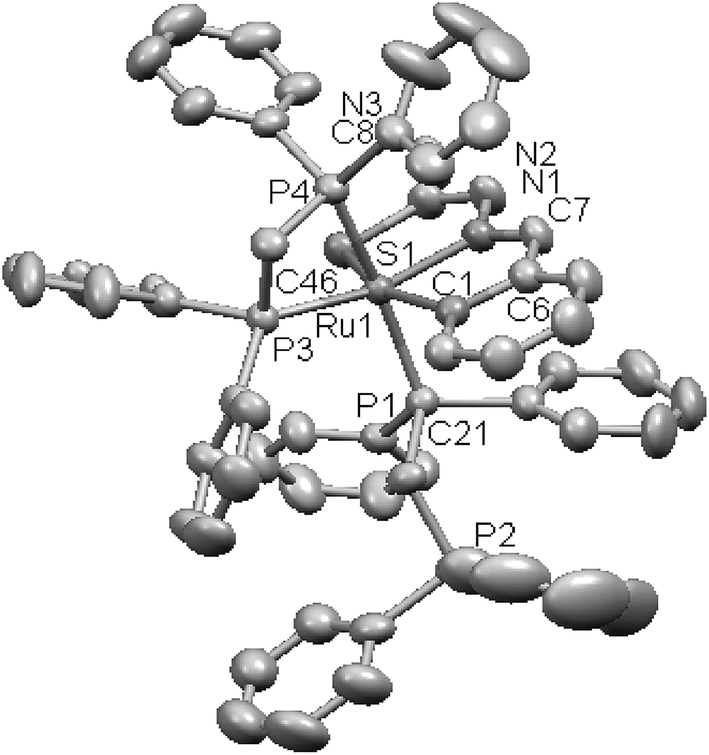
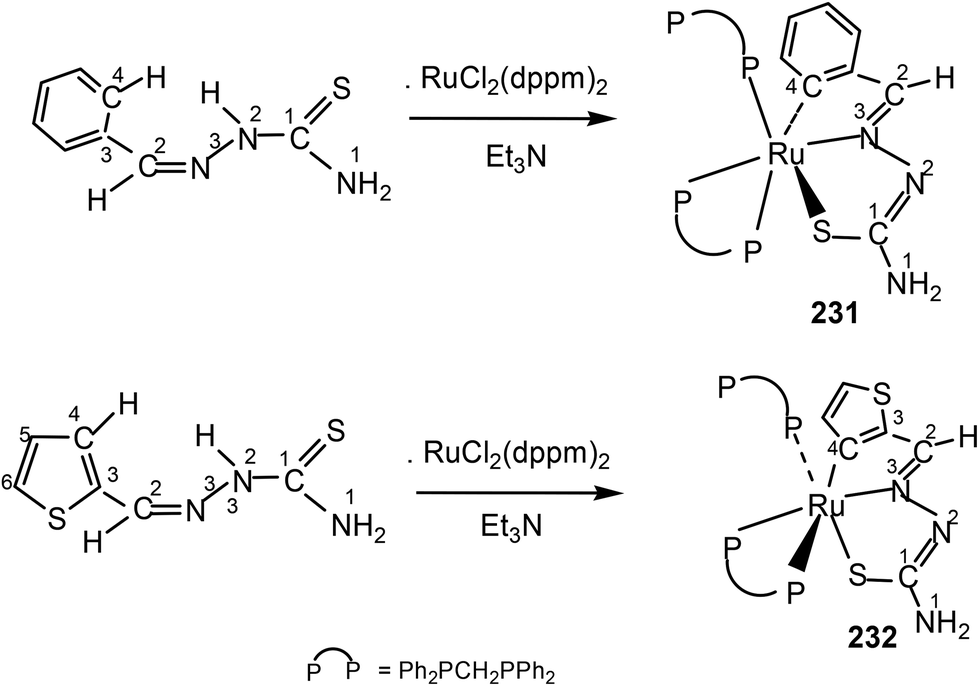
Thiophene-2-carbaldehyde thiosemicarbazone (L88) with trans-Ru(dppm)2Cl2 (dppm = Ph2P–CH2–PPh2) in 2:1 molar ratio (L:M) in toluene in presence of Et3N amine base yielded a complex [Ru(C,N,S-L88-2H)(P,P-dppm) (P-dppm)] 231 – first case of cyclometallation in RuII – thiosemicarbazone chemistry.122 Benzaldehyde thiosemicarbazone (L89) showed similar behavior and formed complex, [Ru(C,N,S-L89-2H)(P,P-dppm)(P-dppm)] 232. Here cyclometallation has been attributed to the short bite of chelating dppm, providing a chance to thio-ligand to come in close proximity of the metal leading to cyclometallation and sixth site is occupied by one end of dppm ligand. No cyclometallation occurred when triphenyl phosphine or tri-p-tolyl phosphine were used in place of dppm in the starting precursor.
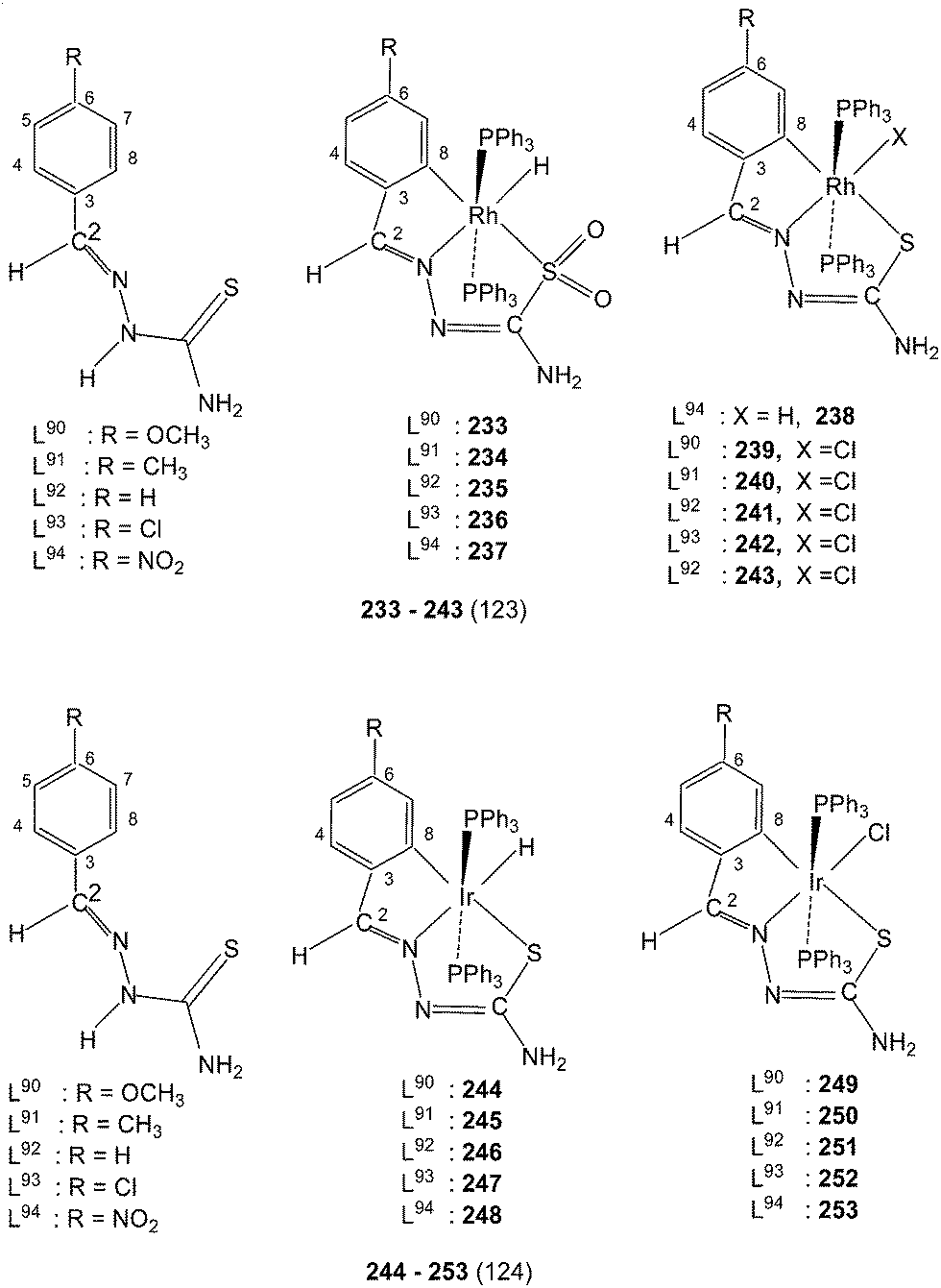
Rhodium and iridium. From the reactions of Rh(PPh3)3Cl with the thio-ligands L90–94 in ethanol in presence of triethyl amine, cyclometallated products 233–237 have been isolated.123 Complexes 237 and 238 were two products from the same ligand L94. Here in 233–237, the coordinated S is oxidized. In all these complexes the ligands are C,N,S donor atoms coordinating to the metal center, the metal is oxidized to RhIII, two positions are occupied by PPh3 ligands and 6th site is occupied by hydride. Cyclometallated complexes 239–243 were obtained similarly but no Et3N base was used in the formation of these complexes.123 In these complexes one site is occupied by chloride ion. Molecular structures of 236, 238 and 242 have been obtained. Reactions of the thio-ligands L90–94 with Ir(PPh3)3Cl (1:1 molar ratio) in ethanol in presence of triethyl amine under dinitrogen atmosphere yielded a series of cylcometallated complexes, [Ir(C,N,S-L)(PPh3)2(H)] 244–248.124 When same reactions were carried out in toluene, two types of cyclometallated complexes 244–248 and [Ir(C,N,S-L)(PPh3)2(Cl)] 249–253 were formed.
3.2 Molecular structures
The molecular structures of several cyclometallated compounds of thiosemicarbazones have been reported and a list of compounds with some important bond parameters is placed in Table 1. Broadly speaking there are four types of cyclometallated compounds of thiosemicarbazones: tetranuclear, trinucelar, dinuclear and mononuclear. Palladium is the only metal which has yielded all the four types of compounds, while platinum gave tetranuclear, dinuclear and mononuclear complexes. Finally, other metals (Ru, Rh, Ir) gave dinuclear (Ru) or mononuclear (Ru, Rh, Ir) compounds. Generally tertiary phosphines appear as the preferred choice (or suitable one) of co-ligands, though a few other co-ligands such as, CO, H− do also appear in the compounds. Only selected original molecular structures are given in this section and briefly commented about their bonding patterns. Further, the bond parameters (Table 1) for all the compounds are also briefly commented in this section.
Table 1 Important bond lengths (Å) and bond angles (°) of complexes
| Palladium(II) |
| Bridging. tsc = thiosemicarbazone ligand. |
| Tri- and tetra-nuclear complexes |
| {Pd3(C,N,μ-S-L11-2H)3} 14 (ref. 99) |
| Pd–C |
2.038(11) |
N3–Pd–S (trans) |
176.5(3) |
| Pd–N3 |
2.001(11) |
C–Pd–S (trans) |
165.9(4) |
| Pd–Sa |
2.403(3), 2.322(4) |
C–Pd–N3 |
86.4(5) |
| S–C |
1.782(13) |
S–Pd–N3 |
82.7(3) |
| {Pd4(C,N,μ-S-L10-2H)4} 10 (ref. 98) |
| Pd–C |
2.021(5) |
N3–Pd–S (trans) |
173.23(12) |
| Pd–N3 |
2.064(4) |
C–Pd–S (trans) |
176.29(14) |
| Pd–Sa |
2.3208(13), 2.3380(14) |
C–Pd–N3 |
94.27(18) |
| S–C |
1.749(6) |
S–Pd–N3 |
83.45(12) |
| {Pd4(C,N,μ-S-L4-2H)4} 4 (ref. 96) |
| Pd–C |
2.01(2) |
N3–Pd–S (trans) |
— |
| Pd–N3 |
1.98(2) |
C–Pd–S (trans) |
— |
| Pd–Sa |
2.382(6), 2.316(5) |
C–Pd–N3 |
80.4(8) |
| S–C |
1.78(2) |
S–Pd–N3 |
81.4(5) |
| {Pd4(C,N,μ-S-L24-2H)4} 27 (ref. 102) |
| Pd–C |
2.015(8) |
N3–Pd–S (trans) |
176.35(18) |
| Pd–N3 |
1.994(7) |
C–Pd–S (trans) |
162.6(3) |
| Pd–Sa |
2.319(2), 2.3611(19) |
C–Pd–N3 |
81.0(3) |
| S–C |
1.790(8) |
S–Pd–N3 |
82.66(18) |
| {Pd4(C,N,μ-S-L27-2H)4} 50 (ref. 103) |
| Pd–C |
2.007(5) |
N3–Pd–S (trans) |
177.11(13) |
| Pd–N3 |
1.989(4) |
C–Pd–S (trans) |
164.66(17) |
| Pd–Sa |
2.3186(16), 2.3551(16) |
C–Pd–N3 |
80.9(2) |
| S–C |
1.792(6) |
S–Pd–N3 |
84.10(13) |
| {Pd4(C,N,μ-S-L30-2H)4} 53 (ref. 103) |
| Pd–C |
1.990(7) |
N3–Pd–S (trans) |
177.48(17) |
| Pd–N3 |
1.988(6) |
C–Pd–S (trans) |
165.0(2) |
| Pd–Sa |
2.315(2), 2.355(2) |
C–Pd–N3 |
81.4(3) |
| S–C |
1.794(7) |
S–Pd–N3 |
84.01(19) |
| {Pd4(C,N,μ-S-L32-2H)4} 55 (ref. 108) |
| Pd–C |
2.023(11) |
N3–Pd–S (trans) |
177.0(3) |
| Pd–N3 |
1.985(10) |
C–Pd–S (trans) |
163.6(4) |
| Pd–S |
2.325(4); 2.369(3) |
C–Pd–N3 |
80.6(5) |
| S–C |
1.747(17) |
S–Pd–N3 |
83.9(3) |
| {Pd4(C,N,μ-S-L36-2H)4} 100 (ref. 106) |
| Pd–C |
2.006(5) |
N3–Pd–S (trans) |
175.78(14) |
| Pd–N3 |
2.003(5) |
C–Pd–S (trans) |
162.97(18) |
| Pd–S |
2.3664(16); 2.2997(16) |
C–Pd–N3 |
81.3(2) |
| S–C |
1.821(6) |
S–Pd–N3 |
82.79(14) |
| {Pd4(C,N,μ-S-L38-2H)4} 102 (ref. 106) |
| Pd–C |
1.979(9) |
N3–Pd–S (trans) |
176.6(2) |
| Pd–N3 |
1.993(7) |
C–Pd–S (trans) |
160.3(3) |
| Pd–S |
2.353(2); 2.317(2) |
C–Pd–N3 |
80.9(4) |
| S–C |
1.773(9) |
S–Pd–N3 |
81.9(2) |
|
| Mono- and di-nuclear complexes |
| Pd(C,N,S-L15-2H)(PPh3)} 18 (ref. 101) |
| Pd–C |
2.051(3) |
N3–Pd–P (trans) |
176.76(9) |
| Pd–N3 |
2.029(2) |
C–Pd–S (trans) |
162.87(7) |
| Pd–S |
2.3560(10) |
C–Pd–N3 |
81.57(10) |
| Pd–P |
2.2498(6) |
S–Pd–N3 |
81.36(9) |
| S–C |
1.754(3) |
|
|
| [Pd(C,N,S-L20-2H)(PPh3)] 30 (ref. 100) |
| Pd–C |
2.044(4) |
N3–Pd–P (trans) |
176.93(9) |
| Pd–N3 |
2.024(3) |
C–Pd–S (trans) |
163.74(10) |
| Pd–S |
2.3419(11) |
C–Pd–N3 |
81.03(13) |
| Pd–P |
2.2519(9) |
S–Pd–N3 |
82.77(9) |
| S–C |
1.757(4) |
|
|
| [Pd(C,N,S-L24-2H)(PPh3)] 34 (ref. 102) |
| Pd–C |
2.033(5) |
N3–Pd–P (trans) |
178.75(11) |
| Pd–N3 |
2.029(4) |
C–Pd–S (trans) |
164.5(2) |
| Pd–S |
2.3270(14) |
C–Pd–N3 |
81.3(2) |
| Pd–P |
2.2527(13) |
S–Pd–N3 |
83.19(11) |
| S–C |
1.750(5) |
|
|
| [Pd(C,N,S-L29-2H)(η1-dppm)] 85 (ref. 103) |
| Pd–C |
2.026(3) |
N3–Pd–P (trans) |
177.29(8) |
| Pd–N3 |
2.018(2) |
C–Pd–S (trans) |
163.95(9) |
| Pd–S |
2.3192(9) |
C–Pd–N3 |
80.77(7) |
| Pd–P |
2.2626(8) |
S–Pd–N3 |
83.23(7) |
| S–C |
1.754(3) |
|
|
| [Pd(C,N,S-L30-2H)(η1-dppm)] 86 (ref. 103) |
| Pd–C |
2.022(4) |
N3–Pd–P (trans) |
177.26(9) |
| Pd–N3 |
2.015(3) |
C–Pd–S (trans) |
164.05(12) |
| Pd–S |
2.3231(11) |
C–Pd–N3 |
80.54(14) |
| Pd–P |
2.2659(10) |
S–Pd–N3 |
83.51(9) |
| S–C |
1.746(4) |
|
|
| [Pd(C,N,S-L32-2H)(η1-P-dppm)] 88 (ref. 108) |
| Pd–C |
2.035(3) |
N3–Pd–P (trans) |
177.75(8) |
| Pd–N3 |
2.034(3) |
C–Pd–S (trans) |
163.81(10) |
| Pd–S |
2.3204(10) |
C–Pd–N3 |
81.21(12) |
| Pd–P |
2.2592(8) |
S–Pd–N3 |
82.75(8) |
| S–C |
1.753(4) |
|
|
| [Pd(C,N,S-L33-2H)(η1-P-dppm)] 89 (ref. 108) |
| Pd–C |
2.038(5) |
N3–Pd–P (trans) |
177.45(12) |
| Pd–N3 |
2.031(4) |
C–Pd–S (trans) |
163.80(15) |
| Pd–S |
2.3323(15) |
C–Pd–N3 |
81.30(19) |
| Pd–P |
2.2616(13) |
S–Pd–N3 |
82.56(12) |
| S–C |
1.758(6) |
|
|
| [Pd(C,N,S-L32-2H)(η1-P-dppen)] 90 (ref. 108) |
| Pd–C |
2.088(2) |
N3–Pd–P (trans) |
179.07(8) |
| Pd–N3 |
2.019(2) |
C–Pd–S (trans) |
163.47(9) |
| Pd–S |
2.3430(13) |
C–Pd–N3 |
80.88(12) |
| Pd–P |
2.2536(10) |
S–Pd–N3 |
82.78(8) |
| S–C |
1.742(3) |
|
|
| [Pd(C,N,S-L40-2H)(Ph3P)] 128 (ref. 109) |
| Pd–C |
2.038(4) |
N3–Pd–P (trans) |
n/a |
| Pd–N3 |
2.036(3) |
C–Pd–S (trans) |
n/a |
| Pd–S |
2.3324(9) |
C–Pd–N3 |
81.30(12) |
| Pd–P |
2.2614(9) |
S–Pd–N3 |
82.48(8) |
| S–C |
1.762(4) |
|
|
| [Pd(C,N,S-L49-2H)(PPh3)] 152 (ref. 111) |
| Pd–C |
2.0411(19) |
N3–Pd–P (trans) |
177.05(5) |
| Pd–N3 |
2.0326(15) |
C–Pd–S (trans) |
163.45(6) |
| Pd–S |
2.3464(6) |
C–Pd–N3 |
81.47(7) |
| Pd–P |
2.2513(5) |
S–Pd–N3 |
82.26(5) |
| S–C |
1.758(2) |
|
|
| [Pd(C,N,S-L49-2H)(PPh3)] 152 (ref. 113) |
| Pd–C |
2.047(3) |
N3–Pd–P (trans) |
176.75(7) |
| Pd–N3 |
2.030(2) |
C–Pd–S (trans) |
163.50(7) |
| Pd–S |
2.349(10) |
C–Pd–N3 |
81.26(9) |
| Pd–P |
2.254(8) |
S–Pd–N3 |
82.49(7) |
| S–C |
1.758(3) |
|
|
| [Pd(C,N,S-L50-2H)(PPh3)] 153 (ref. 112) |
| Pd–C |
2.0390(19) |
N3–Pd–P (trans) |
173.54(5) |
| Pd–N3 |
2.0283(16) |
C–Pd–S (trans) |
164.27(5) |
| Pd–S |
2.3347(5) |
C–Pd–N3 |
81.15(7) |
| Pd–P |
2.2489(5) |
S–Pd–N3 |
83.18(5) |
| [Pd(C,N,S-L51-2H)(PPh3)] 154 (ref. 112) |
| Pd–C |
2.036(4), 2.103(2) |
N3–Pd–P (trans) |
176.69(9) |
| Pd–N3 |
2.038(3), 2.058(3) |
C–Pd–S (trans) |
164.02(11) |
| Pd–S |
2.3225(9), 2.3014(10) |
C–Pd–N3 |
81.41(13) |
| Pd–P |
2.2592(9), 2.2554(10) |
S–Pd–N3 |
82.80(9) |
| [Pd(C,N,S-L52-2H)(PPh3)] 155 (ref. 112) |
| Pd–C |
2.3335(7) |
N3–Pd–P (trans) |
178.44(5) |
| Pd–N3 |
2.0285(17) |
C–Pd–S (trans) |
164.35(6) |
| Pd–S |
2.3335(7) |
C–Pd–N3 |
81.39(8) |
| Pd–P |
2.2466(6) |
S–Pd–N3 |
83.41(5) |
| [Pd(C,N,S-L54-2H)(PPh3)] 157 (ref. 113) |
| Pd–C |
2.027(3) |
N3–Pd–P (trans) |
177.00(8) |
| Pd–N3 |
2.034(3) |
C–Pd–S (trans) |
163.53(8) |
| Pd–S |
2.345(9) |
C–Pd–N3 |
81.37(11) |
| Pd–P |
2.260(8) |
S–Pd–N3 |
82.26(8) |
| S–C |
1.750(3) |
|
|
| [Pd(C,N,S-L56-2H)(PPh3)] 159 (ref. 112) |
| Pd–C |
2.026(5) |
N3–Pd–P (trans) |
173.85(13) |
| Pd–N3 |
2.034(4) |
C–Pd–S (trans) |
163.78(16) |
| Pd–S |
2.3237(15) |
C–Pd–N3 |
81.5(2) |
| Pd–P |
2.2417(14) |
S–Pd–N3 |
82.34(14) |
| [Pd(C,N,S-L57-2H)(PPh3)] 160 (ref. 112) |
| Pd–C |
2.050(14), 2.036(12) |
N3–Pd–P (trans) |
173.7(3), 176.1(4) |
| Pd–N3 |
2.045(13), 2.029(14) |
C–Pd–S (trans) |
165.4(4), 161.5(4) |
| Pd–S |
2.333(4), 2.320(4) |
C–Pd–N3 |
82.1(5), 80.4(5) |
| Pd–P |
2.239(4), 2.241(4) |
S–Pd–N3 |
83.4(4), 81.3(4) |
| {Pd2(C,N,S-L24-2H)2}(μ-Ph2P–CH2–PPh2) 44 (ref. 102) |
| Pd–C |
2.046(7) |
N3–Pd–P (trans) |
178.31(18) |
| Pd–N3 |
2.028(7) |
C–Pd–S (trans) |
163.3(3) |
| Pd–S |
2.348(2) |
C–Pd–N3 |
81.5(3) |
| Pd–P |
2.253(2) |
S–Pd–N3 |
82.27(19) |
| S–C |
1.766(9) |
|
|
| {Pd2(C,N,S-L28-2H)2}(μ-Ph2P–CH2–CH2–CH2–PPh2) 62 (ref. 105) |
| Pd–C |
2.040(10) |
N3–Pd–P (trans) |
177.4(2) |
| Pd–N3 |
2.020(7) |
C–Pd–S (trans) |
163.7(2) |
| Pd–S |
2.322(3) |
C–Pd–N3 |
80.6(3) |
| Pd–P |
2.274(2) |
S–Pd–N3 |
83.7(2) |
| S–C |
1.743(10) |
|
|
| {Pd(C,N,S-L31-2H)}2(μ-P,P-dppe) 67 (ref. 106) |
| Pd–C |
2.047(7) |
N3–Pd–P (trans) |
175.97(16) |
| Pd–N3 |
2.008(5) |
C–Pd–S (trans) |
163.53(18) |
| Pd–S |
2.3209(19) |
C–Pd–N3 |
80.2(2) |
| Pd–P |
2.2637(18) |
S–Pd–N3 |
83.32(16) |
| S–C |
1.743(7) |
|
|
| {Pd(C,N,S-L30-2H)}2(μ-P,P-dppe) 72 (ref. 108) |
| Pd–C |
2.042(6) |
N3–Pd–P (trans) |
176.03(16) |
| Pd–N3 |
2.025(5) |
C–Pd–S (trans) |
163.78(17) |
| Pd–S |
2.3225(19) |
C–Pd–N3 |
80.4(2) |
| Pd–P |
2.2544(16) |
S–Pd–N3 |
83.69(16) |
| S–C |
1.766(8) |
|
|
| {Pd(C,N,S-L26-2H)}2(μ-P,P-dppen) 75 (ref. 108) |
| Pd–C |
2.023(12) |
N3–Pd–P (trans) |
178.7(3) |
| Pd–N3 |
2.012(9) |
C–Pd–S (trans) |
164.7 (3) |
| Pd–S |
2.327(4) |
C–Pd–N3 |
81.0(4) |
| Pd–P |
2.259(3) |
S–Pd–N3 |
83.6(3) |
| S–C |
1.764(12) |
|
|
| {Pd(C,N,S-L32-2H)}2(μ-P,P-dppen) 76 (ref. 108) |
| Pd–C |
2.036(2) |
N3–Pd–P (trans) |
178.77(6) |
| Pd–N3 |
2.022(2) |
C–Pd–S (trans) |
164.51(8) |
| Pd–S |
2.3208(8) |
C–Pd–N3 |
81.26(9) |
| Pd–P |
2.2421(7) |
S–Pd–N3 |
83.25(6) |
| S–C |
1.759(3) |
|
|
| {Pd(C,N,S-L34-2H)}2(μ-P,P-dppe) 79 (ref. 107) |
| Pd–C |
2.0324(2) |
N3–Pd–P (trans) |
176.18(8) |
| Pd–N3 |
2.024(3) |
C–Pd–S (trans) |
163.42(3) |
| Pd–S |
2.3333(9) |
C–Pd–N3 |
80.55(8) |
| Pd–P |
2.2663(8) |
S–Pd–N3 |
82.88(8) |
| S–C |
1.7704(11) |
|
|
| {Pd(C,N,S-L35-2H)}2(μ-P,P-dppp) 105 (ref. 105) |
| Pd–C |
2.044(8) |
N3–Pd–P (trans) |
177.67(18) |
| Pd–N3 |
2.039(6) |
C–Pd–S (trans) |
162.8(8) |
| Pd–S |
2.337(2) |
C–Pd–N3 |
80.6(3) |
| Pd–P |
2.261(2) |
S–Pd–N3 |
82.22(18) |
| S–C |
1.769(8) |
|
|
| [Pd2Cl2(C,N,μ-S-L32-2H)(μ-P,P-dppm)] 97 (ref. 104) |
| Pd1–C |
2.012(4) |
N3–Pd(1)–P (trans) |
176.65(10) |
| Pd1–N3 |
2.020(3) |
C–Pd(1)–S (trans) |
165.15(12) |
| Pd1–S |
2.3214(11) |
C–Pd(1)–N3 |
81.16(16) |
| Pd–P |
2.2465(11) |
S–Pd(1)–N3 |
84.00(10) |
| S–C |
n/a |
|
|
| Pd2–S (bridging) |
2.3074(11) |
S(1)–Pd(2)–Cl(1) |
170.41(4) |
| Pd2–P |
2.2644(11) |
P(2)–Pd(2)–Cl(2) |
179.20(4) |
| Pd2–Cl |
2.3160(11), 2.3448(11) |
|
|
| [Pd2Cl2(C,N,μ-S-L46-2H)(μ-P,P-bdcp)] 141 (ref. 110) |
| Pd1–C |
2.025(4) |
N3–Pd–P (trans) |
— |
| Pd1–N3 |
2.023(3) |
C–Pd–S (trans) |
— |
| Pd1–S |
2.3276(11) |
C–Pd–N3 |
80.64(15) |
| Pd–P |
2.2579(12) |
S–Pd–N3 |
83.42(10) |
| S–C |
1.810(4) |
|
|
| Pd2–S (bridging) |
2.2943(12) |
Cl–Pd(2)–Cl |
88.14(4) |
| Pd2–P |
2.2624(12) |
S–Pd–P(2) |
90.01(4) |
| Pd2–Cl |
2.3415(13); 2.3288(12) |
|
|
| [Pd2(C,N,μ-S-L41-2H)(μ-P,P-dppm)(κ2-P,P-dppm)](ClO4)2 143 (ref. 110) |
| Pd1–C |
2.032(3) |
N3–Pd–P (trans) |
— |
| Pd1–N3 |
2.029(2) |
C–Pd–S (trans) |
— |
| Pd1–S |
2.3361(11) |
C–Pd(1)–N3 |
81.13(11) |
| Pd–P |
2.2553(8) |
S–Pd(1)–N3 |
82.31(8) |
| S–C |
1.807(3) |
S(1)–Pd(2)–P(2) |
83.65(3) |
| Pd2–S (bridging) |
2.3627(8) |
P(3)–Pd(2)–P(4) |
70.56(3) |
| Pd2–P |
2.3452(8) |
|
|
| [Pd2(C,N,μ-S-L44-2H)(μ-P,P-bdcp) (κ2-P,P-dppm)](ClO4)2 147 (ref. 110) |
| Pd1–C |
2.038(6) |
N3–Pd–P (trans) |
— |
| Pd1–N3 |
2.034(5) |
C–Pd–S (trans) |
— |
| Pd1–S |
2.3214(16) |
C–Pd(1)–N3 |
80.5(2) |
| Pd1–P |
2.2520(17) |
S–Pd(1)–N3 |
83.24(14) |
| S–C |
1.810(6) |
S(1)–Pd(2)–P(2) 8 |
87.30(6) |
| Pd2–S (bridging) |
2.3402(18) |
P(3)–Pd(2)–P(4) |
69.87(6) |
| Pd2–P |
2.3515(17) |
|
|
|
| Platinum(II) and ruthenium(II) complexes |
| {Pt4(C,N,μ-S-L4-2H)4} 182 (ref. 96) |
| Pt–C |
2.020(5) |
N3–Pt–S (trans) |
— |
| Pt–N3 |
1.996(5) |
C–Pt–S (trans) |
— |
| Pt–Sa |
2.3491(16), 2.3036(15) |
C–Pt–N3 |
83.57(15) |
| S–C |
1.798(5) |
S–Pt–N3 |
80.8(2) |
| [Pt(C,N,S-L60-2H)(Ph3P)] 170 (ref. 115) |
| Pt–C |
2.02(2) |
N3–Pt–P (trans) |
176.0(4) |
| Pt–N3 |
2.03(2) |
C–Pt–S (trans) |
161.4(5) |
| Pt–S |
2.335(5) |
C–Pt–N3 |
77.9(6) |
| Pt–P |
2.235(5) |
S–Pt–N3 |
83.7(4) |
| S–C |
1.78(2) |
|
|
| [Pt(C,N,S-L65-2H)(Ph3P)] 187 (ref. 116) |
| Pt–C |
2.045(4) |
N3–Pt–P (trans) |
176.87(12) |
| Pt–N3 |
2.032(4) |
C–Pt–S (trans) |
162.52(13) |
| Pt–S |
2.340(13) |
C–Pt–N3 |
80.85(17) |
| Pt–P |
2.230(10) |
S–Pt–N3 |
81.70(13) |
| S–C |
1.755(5) |
|
|
| {Pt2(C,N,S-L70-2H)2}(Ph2P–CH2–CH2–PPh2) 198 (ref. 117) |
| Pt–C |
2.022(5) |
N3–Pt–P (trans) |
n/a |
| Pt–N3 |
2.050(4) |
C–Pt–S (trans) |
n/a |
| Pt–S |
2.317(2) |
C–Pt–N3 |
80.42(2) |
| Pt–P |
2.220(1) |
S–Pt–N3 |
82.00(12) |
| S–C |
1.773(5) |
|
|
| [Ru(C,N3,S-L72-2H)(PPh3)2(CO)] 208 (ref. 118) |
| Ru–C(tsc)/Ru–C(CO)b |
2.077(7)/1.796(8) |
P–Ru–P (trans) |
173.41(7) |
| Ru–N3 |
2.109(6) |
C(tsc)–Ru–S (trans)b |
156.22(19) |
| Ru–S |
2.4614(19) |
C(CO)–Ru–N3 (trans) |
172.5(3) |
| Ru–P |
2.366(2), 2.350(2) |
C–Ru–N3 |
78.9(2) |
| S–C |
1.731(7) |
S–Ru–N3 |
77.35(16) |
| [Ru(C,N,S-L79-2H)(CO)(AsPh3)2] 220 (ref. 120) |
| Ru–C(tsc)/Ru–C(CO)b |
2.061(3)/1.838(3) |
As–Ru–As (trans) |
175.180(13) |
| Ru–N3 |
2.0732(19) |
C(tsc)–Ru–S (trans)b |
157.42(7) |
| Ru–S |
2.4641(7) |
C(CO)–Ru–N3 (trans) |
170.64(11) |
| Ru–As |
2.4695(3), 2.4379(3) |
C–Ru–N3 |
78.41(9) |
| S–C |
1.755(2) |
S–Ru–N3 |
79.05(6) |
| [Ru(C,N,S-L88-2H)(P,P-dppm)(P-dppm)] 231 (ref. 122) |
| Ru–C |
2.076(3) |
P–Ru–P (trans) |
174.36(3) |
| Ru–N3 |
2.067(3) |
C–Ru–S (trans) |
156.92(11) |
| Ru–S |
2.4369(11) |
P–Ru–N3 |
161.92(9) |
| Ru–P |
2.2918(11), 2.3138(11), 2.3684(11) |
C–Ru–N3 |
78.95(13) |
| P–Ru–P (chelating) |
71.33(4) |
S–Ru–N3 |
79.21(8) |
| [Ru(C,N,S-L89-2H)(P,P-dppm)(P-dppm)] 232 (ref. 122) |
| Ru–C |
2.086(3) |
P–Ru–P (trans) |
169.38(3) |
| Ru–N3 |
2.058(2) |
C–Ru–S (trans) |
157.58(8) |
| Ru–S |
2.4385(8) |
P–Ru–N3 |
162.58(7) |
| Ru–P |
2.3384(8), 2.3045(7), 2.3392(8) |
C–Ru–N3 |
78.90(10) |
| P–Ru–P (chelating) |
69.99(3) |
S–Ru–N3 |
79.37(7) |
| [Ru (C,N,S-L87-2H)(CO)(PPh3)2] 228 (ref. 121) |
| Ru–C(tsc)/Ru–C(CO)b |
2.072(8)/1.831(8) |
P–Ru–P (trans) |
176.81(7) |
| Ru–N3 |
2.096(6) |
C(tsc)–Ru–S (trans)b |
158.0(2) |
| Ru–S |
2.437(3) |
C(CO)–Ru–N3 (trans) |
176.7(3) |
| Ru–P |
2.379(3), 2.387(3) |
C–Ru–N3 |
78.3(3) |
| S–C |
1.735(9) |
S–Ru–N3 |
79.83(17) |
| [Ru2(C,N,S-L74-2H)2(PPh3)2(CO)2] 215 (ref. 119) |
| Ru–C(tsc)/Ru–C(CO)b |
2.050(3)/1.857(3) |
P–Ru–S (trans) |
172.34(3) |
| Ru–N3 |
2.092(2) |
C(tsc)–Pt–S (trans)b |
157.19(8) |
| Ru–S |
2.4786(7), 2.4804(7) |
C(CO)–Ru–N3 (trans) |
173.29(11) |
| Ru–P |
2.3071(7) |
C–Ru–N3 |
n/a |
| S–C |
1.781(3) |
S–Ru–N3 |
n/a |
| [RuPt{(C,N,S)2-L87-4H}(CO)(PPh3)3] 230 (ref. 121) |
| Ru–C(tsc)/Ru–C(CO)b |
2.093(8)/1.830(10) |
P–Ru–S (trans) |
171.88(9) |
| Ru–N3 |
2.099(7) |
C(tsc)–Ru–S (trans)b |
157.9(2) |
| Ru–S |
2.462(3) |
C(CO)–Ru–N3 (trans) |
177.2(3) |
| Ru–P |
2.360(3), 2.423(3) |
C–Ru–N3 |
79.0(3) |
| S–C |
1.734(10) |
S–Ru–N3 |
78.98(19) |
| Pt–C |
2.048(9) |
N3–Pt–P (trans) |
174.2(2) |
| Pt–N3 |
2.034(7) |
C–Pt–S (trans) |
162.6(3) |
| Pt–S |
2.339(3) |
C–Pt–N3 |
80.9(3) |
| Pt–P |
2.236(3) |
S–Pt–N3 |
82.0(2) |
| S–C |
1.758(10) |
|
|
|
| Rhodium(III) and iridium(iii): mononuclear complexes |
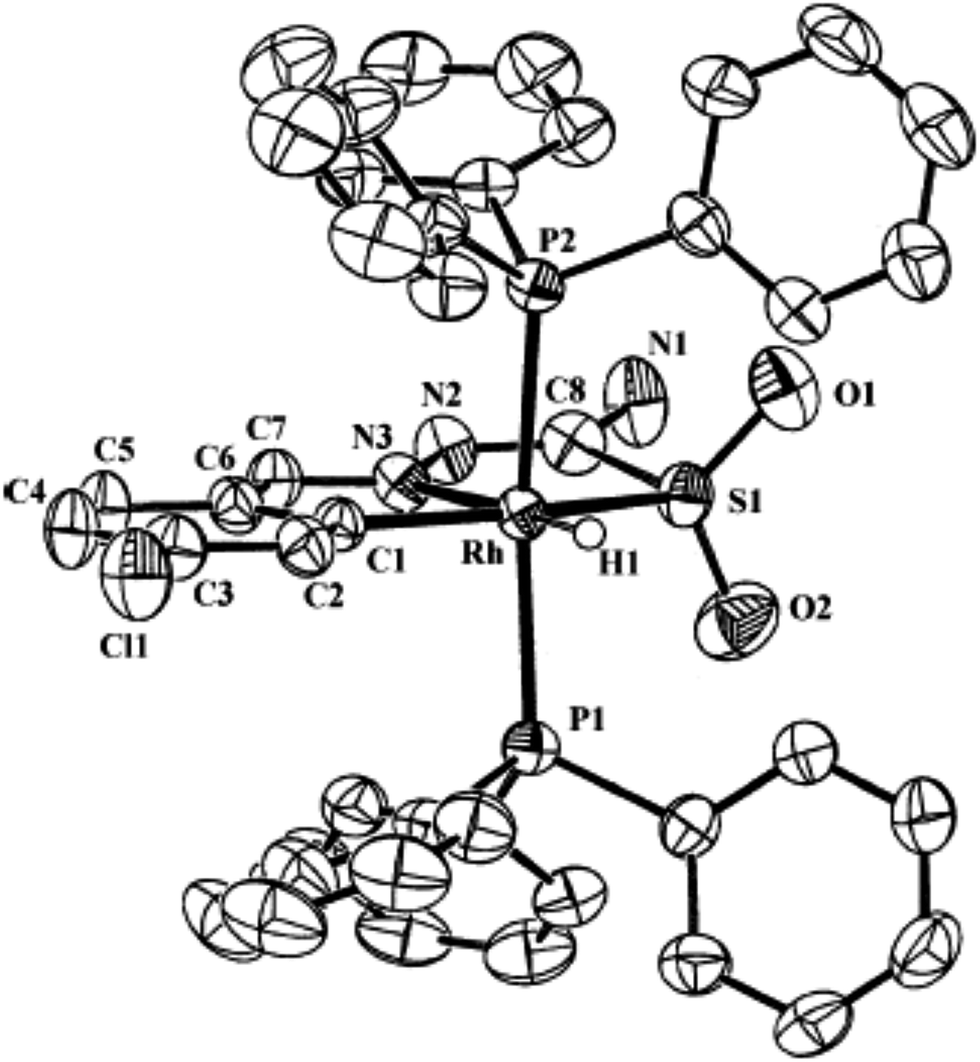
| [Rh {C,N,S(O)2-L93-2H}(H)(PPh3)2] 236 (ref. 123) |
| Rh–C |
2.039(3) |
SO |
1.461(2), 1.465(2) |
| Rh–N3 |
2.096(2) |
P–Rh–P (trans) |
165.40(3) |
| Rh–S |
2.3366(8) |
C–Rh–S (trans) |
160.14(9) |
| Rh–P |
2.3380(8), 2.3483(8) |
H–Rh–N3(trans) |
172.61(7) |
| Rh–H |
0.437(4) |
S–Rh–N3 |
80.46(7) |
| S–C |
1.855(3) |
C–Rh–N3 |
79.73(10) |
| [Rh(C,N,S-L94-2H)(H)(PPh3)2] 238 (ref. 106) |
| Rh–C |
2.038(5) |
P–Rh–P (trans) |
169.23(6) |
| Rh–N3 |
2.074(4) |
C–Rh–S (trans) |
159.99(17) |
| Rh–S |
2.4116(15) |
H–Rh–N3 (trans) |
176(2) |
| Rh–P |
2.3218(16), 2.3041(15) |
S–Rh–N3 |
79.91(12) |
| Rh–H |
1.57(6) |
C–Rh–N3 |
80.2(2) |
| S–C |
1.743(6) |
|
|
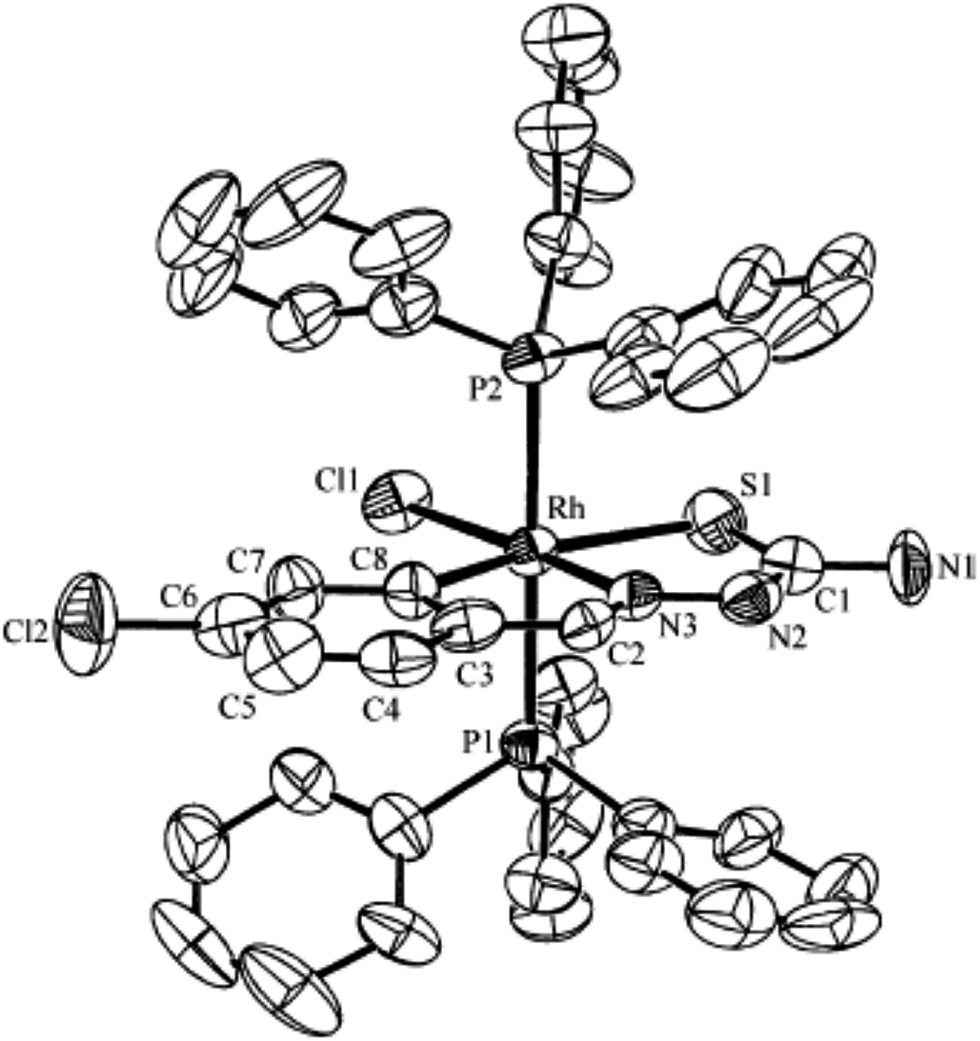
| [Rh (C,N,S-L93-2H)(Cl)(PPh3)2] 242 (ref. 123) |
| Rh–C |
2.044(11) |
P–Rh–P (trans) |
179.56(10) |
| Rh–N3 |
1.990(10) |
C–Rh–S (trans) |
162.2(3) |
| Rh–S |
2.450(3) |
Cl–Rh–N3 (trans) |
176.1(3) |
| Rh–P |
2.379(3), 2.371(3) |
S–Rh–N3 |
81.1(3) |
| Rh–Cl |
2.385(3) |
C–Rh–N3 |
81.1(4) |
| S–C |
1.708(14) |
|
|
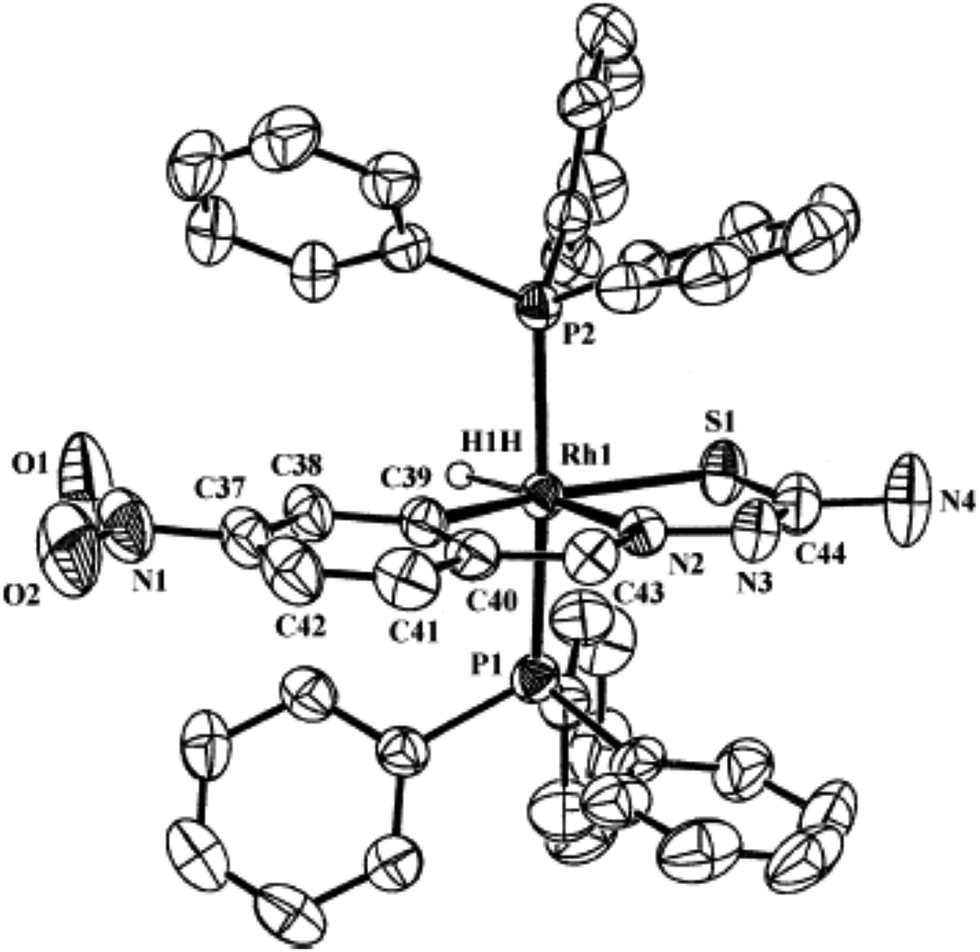
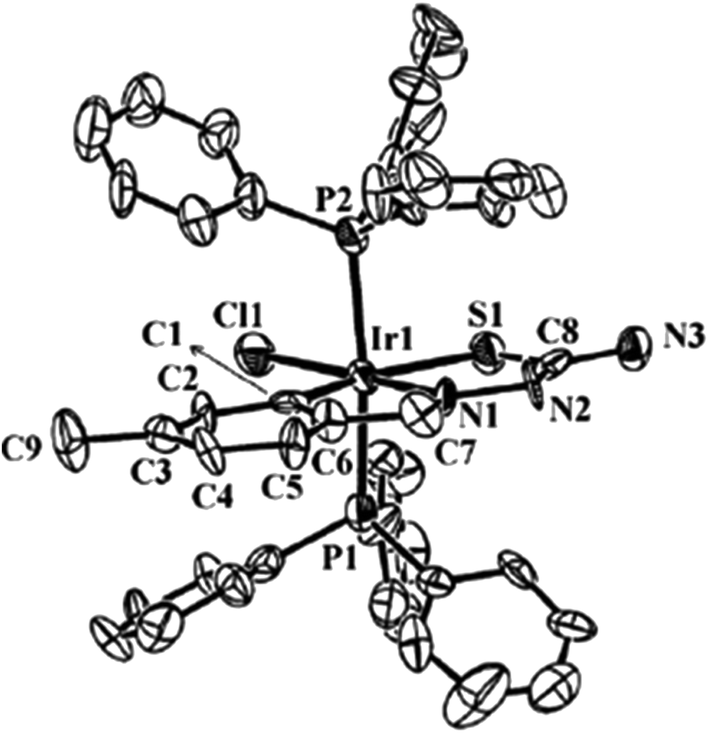
| [Ir(C,N,S-L94-2H)(H)(PPh3)2] 248 (ref. 124) |
| Ir–C |
2.042(8) |
P–Ir–P (trans) |
165.43(8) |
| Ir–N3 |
2.073(7) |
C–Ir–S (trans) |
159.7(2) |
| Ir–S |
2.424(2) |
H–Ir–N3 (trans) |
175(4) |
| Ir–P |
2.302(2), 2.305(2) |
S–Ir–N3 |
79.5(2) |
| Ir–H |
1.41(2) |
C–Ir–N3 |
80.3(3) |
| S–C |
1.738(9) |
|
|
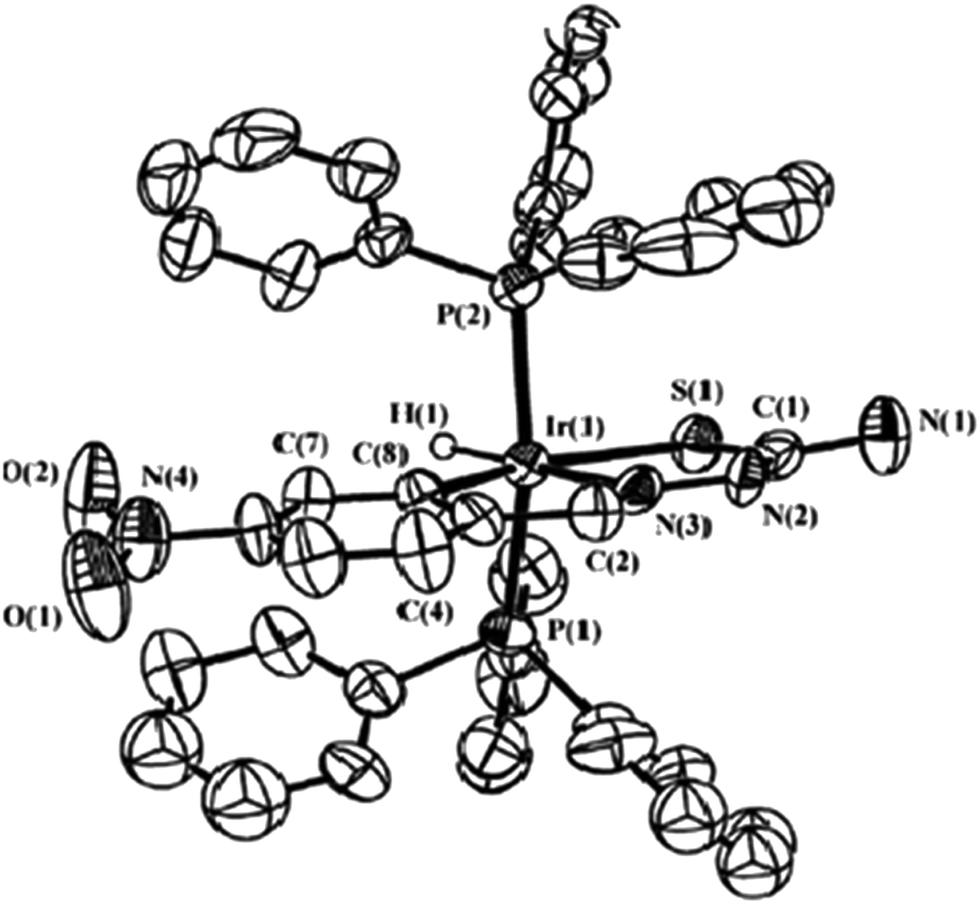
| [Ir(C,N,S-L91-2H)(Cl)(PPh3)2] 250 (ref. 124) |
| Ir–C |
2.013(9) |
P–Ir–P (trans) |
169.53(8), 169.53(8) |
| Ir–N3 |
2.006(7) |
C–Ir–S (trans) |
160.4(2) |
| Ir–S |
2.433(3) |
Cl–Ir–N3 (trans) |
173.7(2) |
| Ir–P |
2.346(3), 2.367(3) |
S–Ir–N3 |
81.1(2) |
| Ir–Cl |
2.389(3) |
C–Ir–N3 |
79.4(3) |
| S–C |
1.745(9) |
|
|
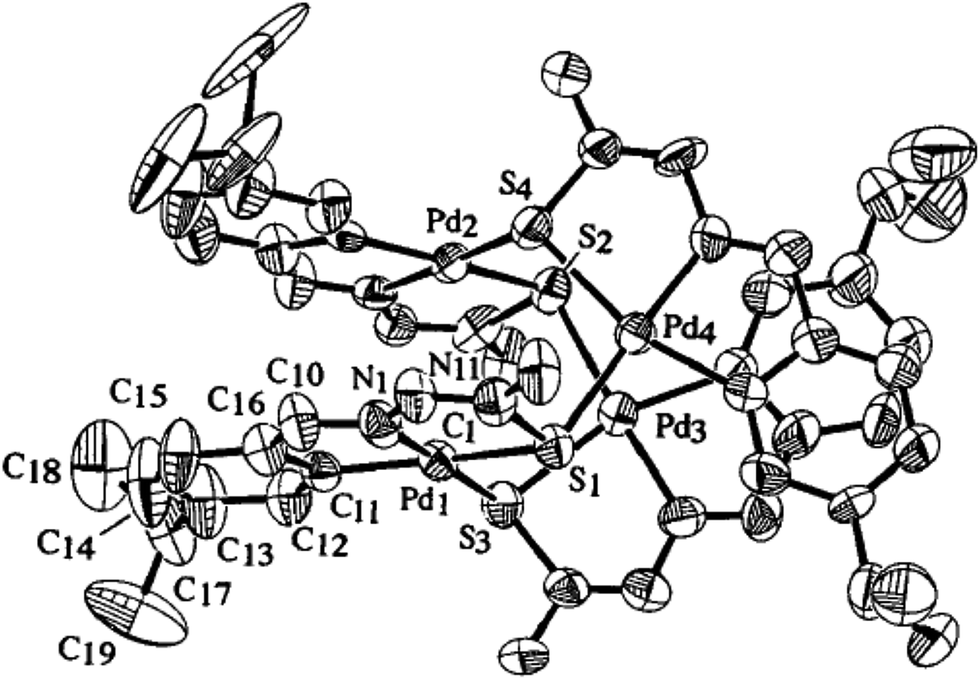
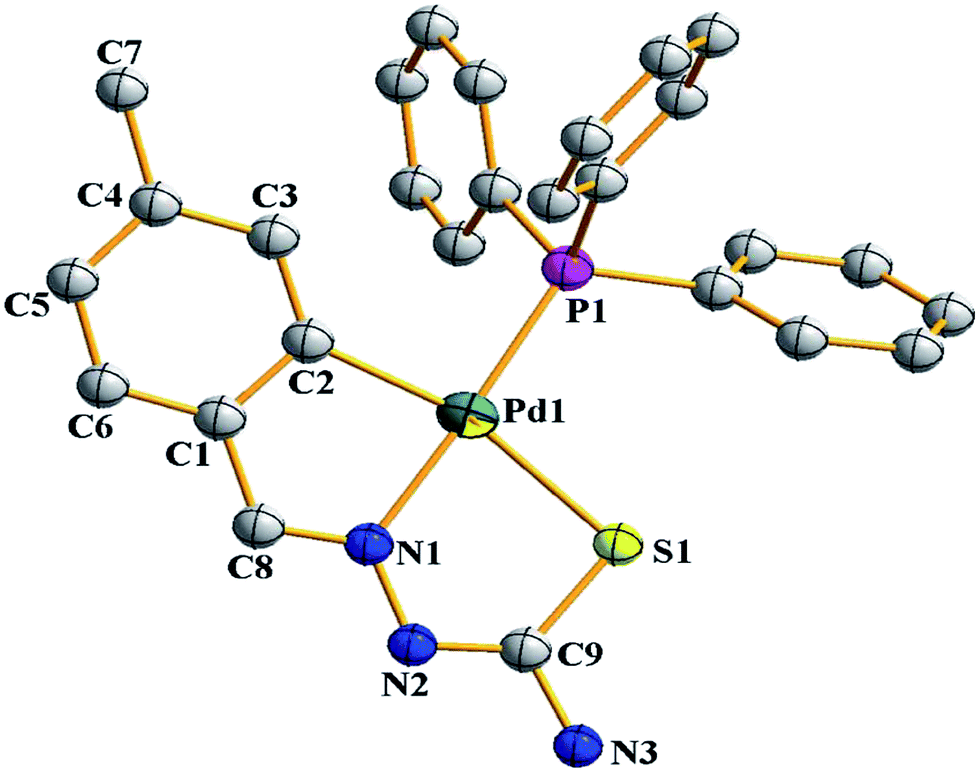
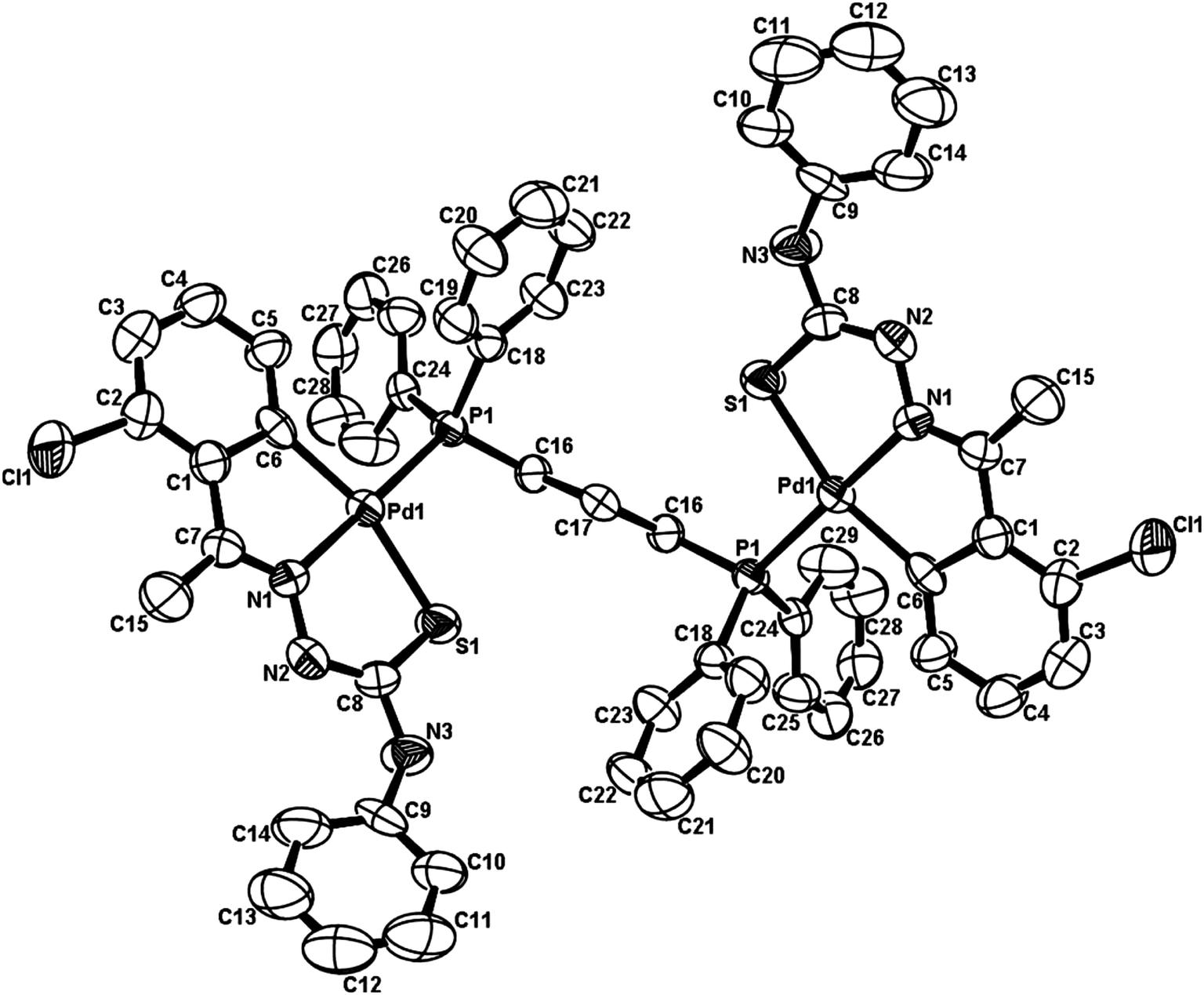
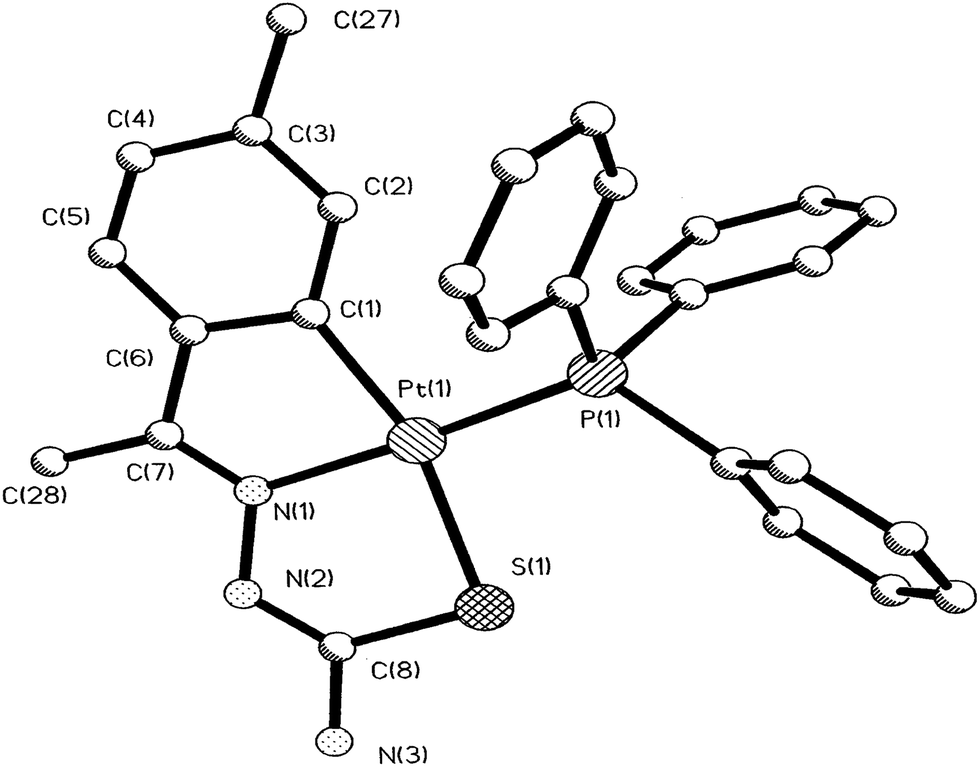
Palladium. There are eight tetra-nuclear compounds (Pd: 4, 10, 27, 50, 53, 55, 100 and 102)96,98,99,102,103,106,108 for which molecular structures are determined (Table 1). All these tetra-nuclear compounds are identical in bonding (C,N,μ-S). Usually there is one substituent in the phenyl ring (ligand types: A, B), though there are cases with more than one substituent as described in Section 3.1. Molecular structures of four representative tetranuclear oligomers 27 (L24), 4 (L4), 100 (L36) and 10 (L10) are shown in Fig. 1–4 respectively. Ligand L24 has no substituent in the phenyl ring (R = H) with R2 = Et at C2 carbon (27, Fig. 1),102 ligand L4 has one substituent in the phenyl ring (R = –CHMe2) with R2 = H at C2 carbon (4, Fig. 2),96 and L36 has substituents in phenyl ring (R = F) and also at C2 (R2 = Me) carbon atoms (100, Fig. 3).106 In these three cases, tetranuclear complexes were formed involving phenyl ring metallation at C8 carbon [ortho to ipso (C3) carbon]. Compounds 4, 27 and 100 belong to category A ligands. The thio-ligand L10 which belongs to category C has also formed tetranuclear compound 10 (Fig. 4). In each case, a thio-ligand as dianion chelates to one metal center and oligomerization occurs through sulfur; each sulfur bridges two metal centers. The central core, Pd4S4 of oligomers forms an eight membered ring. Molecular structure of tri-nuclear complex 14(L11) is shown in Fig. 5.99 Here the thio-ligand L11 has R2 = H, R3 = Et with phenyl ring having two methyl substituents at 4- and 7-positions. It is the methyl group at 4-position which is metallated and not the phenyl ring owing to possibility of its closer proximity to the metal. Thiosemicarbazone (L11) as dianion is coordinating as (C,N,μ-S) and the central core Pd3S3 of the trinuclear complex adopts a non-planar six-membered ring.99
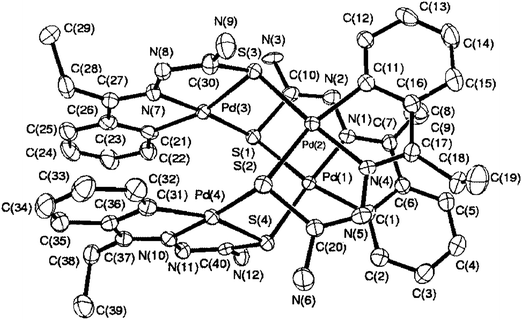 |
| | Fig. 1 Molecular structure of {Pd4(C,N,μ-S-L24-2H)4} 27 (L24: R2 = Et, R3 = H, R = H) [reproduced with permission from ref. 102. Copyright (1999) Royal Society of Chemistry]. | |
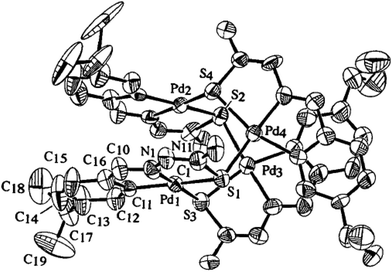 |
| | Fig. 2 Molecular structure of {Pd4(C,N,μ-S-L4-2H)4} 4 (L4: R2, R3 = H, H; R = –CHMe2 at 6-position) [reproduced with permission from ref. 96. Copyright (1998). American Chemical Society]. | |
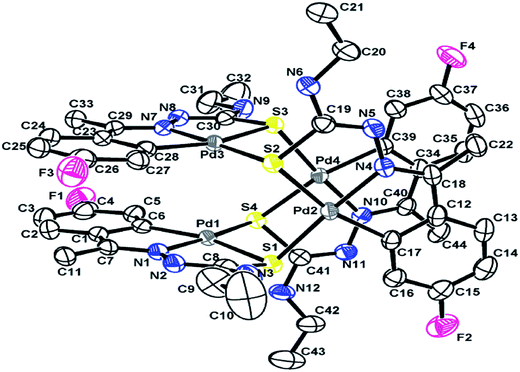 |
| | Fig. 3 Molecular structure of {Pd4(C,N,μ-S-L36-2H)4} 100 (L36: R2 = Me, R3 = Et, R = F at 6-position) [reprinted with permission from ref. 106. Copyright (2012) Elsevier]. | |
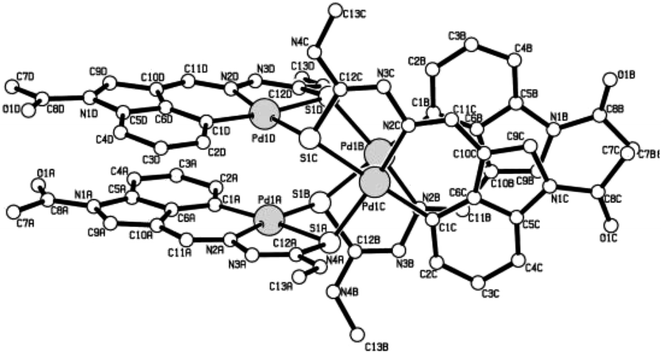 |
| | Fig. 4 Molecular structure of {Pd4(C,N,μ-S-L10-2H)4} 10 (L10: R = Me) [reproduced with permission from ref. 98 Copyright (2006) Wiley-VCH Verlag GmbH & Co. KGaA]. | |
 |
| | Fig. 5 Molecular structure of {Pd3(C,N,μ-S-L11-2H)3} 14 [reprinted with from ref. 99. Copyright (2009) Elsevier]. | |
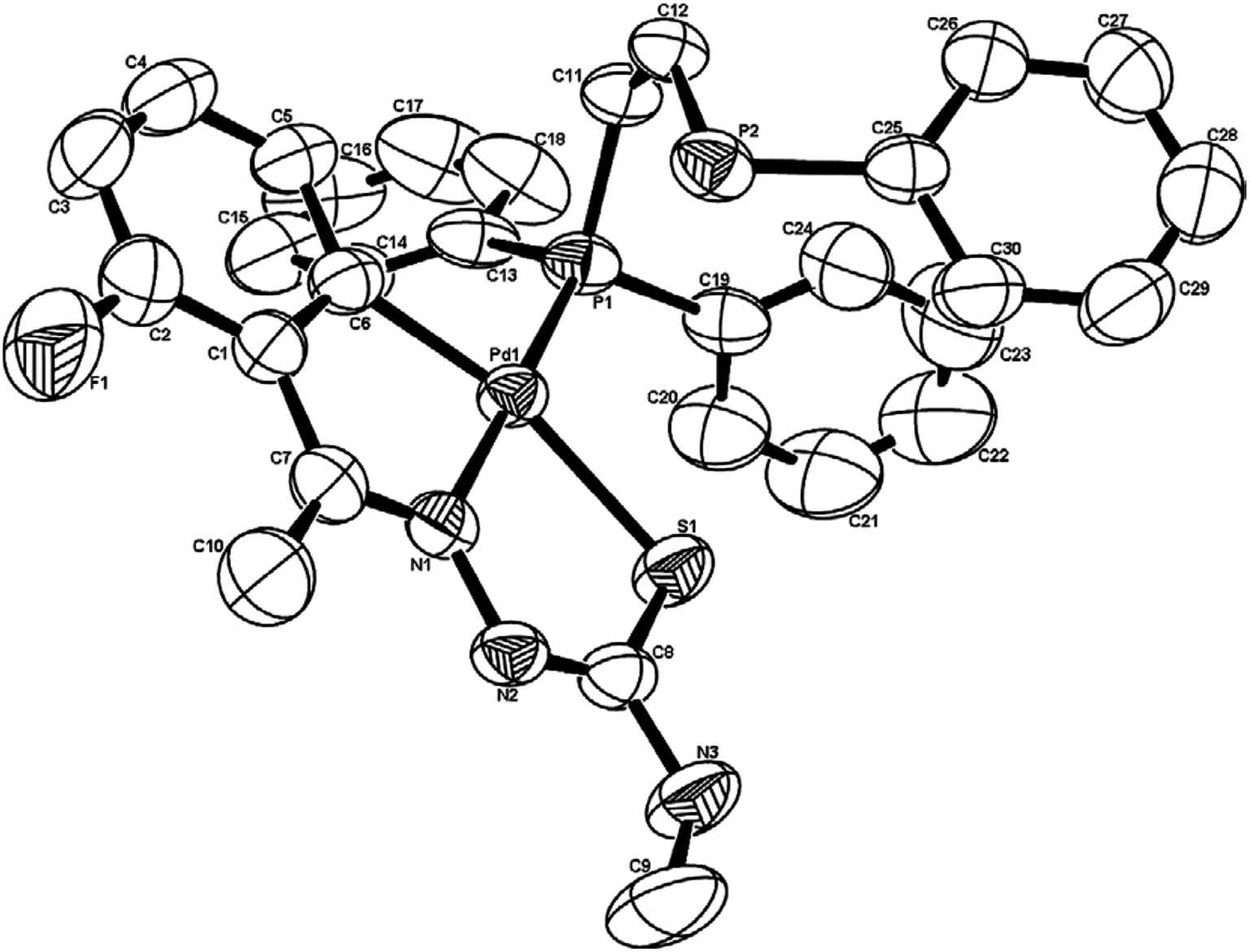
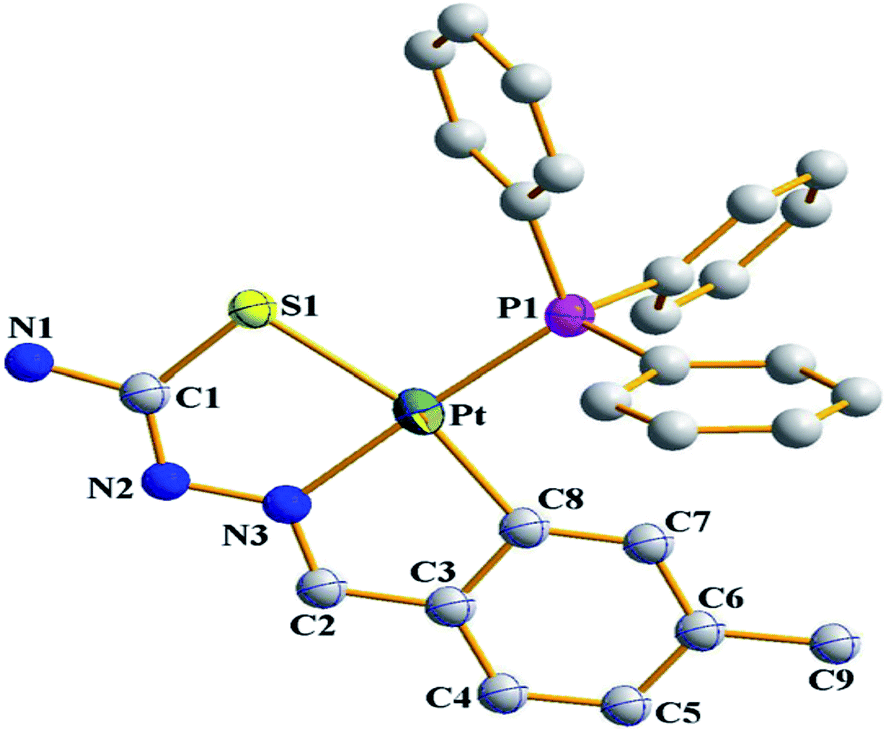
There are seventeen mononuclear (18, 30, 34, 85, 86, 88–90, 128, 152–155, 157, 159, 160)100–103,108,109,111–113 and twelve dinuclear (44, 62, 67, 72, 75, 76, 79, 97, 105, 141, 143, 147)102,104–108,110 cyclopalladated compounds whose molecular structures have been reported (Table 1). These mono- and di-nuclear compounds have terdentate (C,N,S) thio-ligands with PPh3 and bis(tertiaryphosphines) as co-ligands. Bis(tertiaryphosphines) are either mono-dentate (dppm, dppen) or bridging bidentate (dppe, dppen, etc.). Representative molecular structures are depicted below. The thio-ligands L15 and L56 with PPh3 as co-ligand have formed mononuclear four coordinated square planar complexes, [Pd(C,N,S-L15-2H)(PPh3)] 18 (L15: R2 = H, R3 = H, R = Me at 6-position) (Fig. 6) and [Pd(C,N,S-L56-2H)(PPh3)] 159 (L56: R2 = Me, R3 = Me, see ligand type D above) (Fig. 7). In both cases either methyl-substituted phenyl (18) or thiopheneyl) (159) rings are metallated. Bis(tertiaryphosphines), dppm and dppen are mono-denate in four coordinated square planar complexes, [Pd(C,N,S-L29-2H)(η1-P-dppm)] 85 (L29: R2 = Me, R3 = Me, R = Br at 4-position) (Fig. 8) and [Pd(C,N,S-L32)(η1-P-dppen)] 90 (L32: R2 = Me, R3 = Me, R = F at 4-position) (Fig. 9) with one functional group, –PPh2 pendant in both cases.
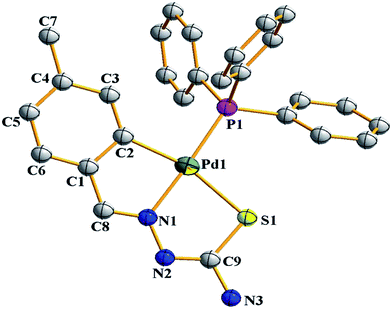 |
| | Fig. 6 Molecular structure of Pd(C,N,S-L15-2H)(PPh3)} 18 (L15: R2 = H, R3 = H, R = Me at 6-position) [reproduced with permission from ref. 101. Copyright (2013) Royal Societyof Chemistry]. | |
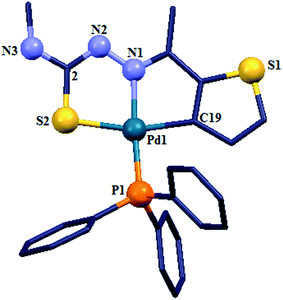 |
| | Fig. 7 Molecular structure of [Pd(C,N,S-L56-2H)(PPh3) ]159 (L56: R2 = Me, R3 = Me, see ligand type D above) [reprinted with permission from ref. 112. Copyright (2012) Elsevier]. | |
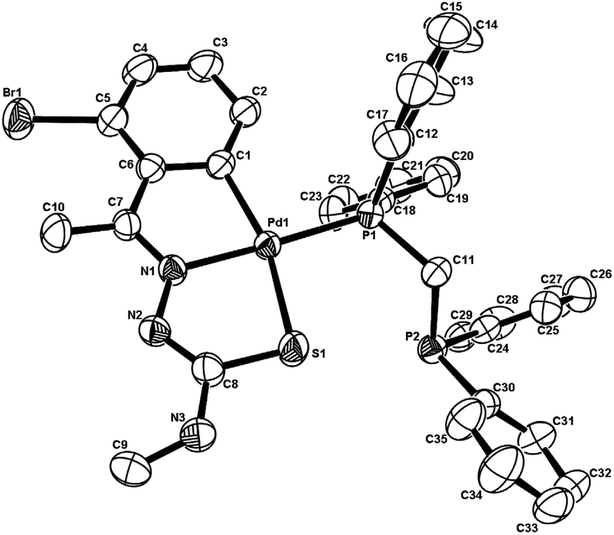 |
| | Fig. 8 Molecular structure of [Pd(C,N,S-L29-2H)(η1-P-dppm)] 85 (L29: R2 = Me, R3 = Me, R = Br at 4-position) [reprinted with permission from ref. 103. Copyright (2006) Elsevier]. | |
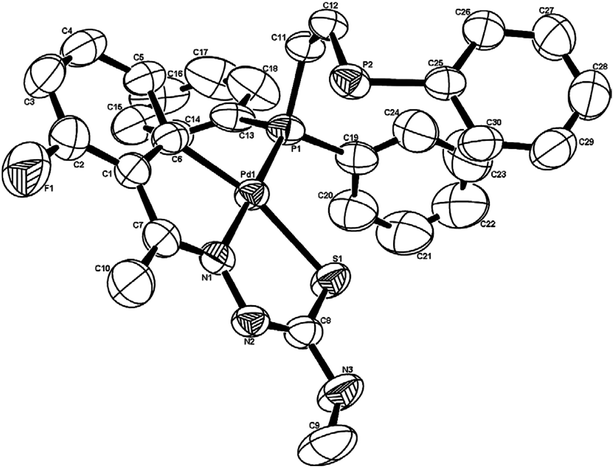 |
| | Fig. 9 Molecular structure of [Pd(C,N,S-L32)(η1-P-dppen)] 90 (L32: R2 = Me, R3 = Me, R = F at 4-position) [reprinted with permission from ref. 108. Copyright (2006) Elsevier]. | |
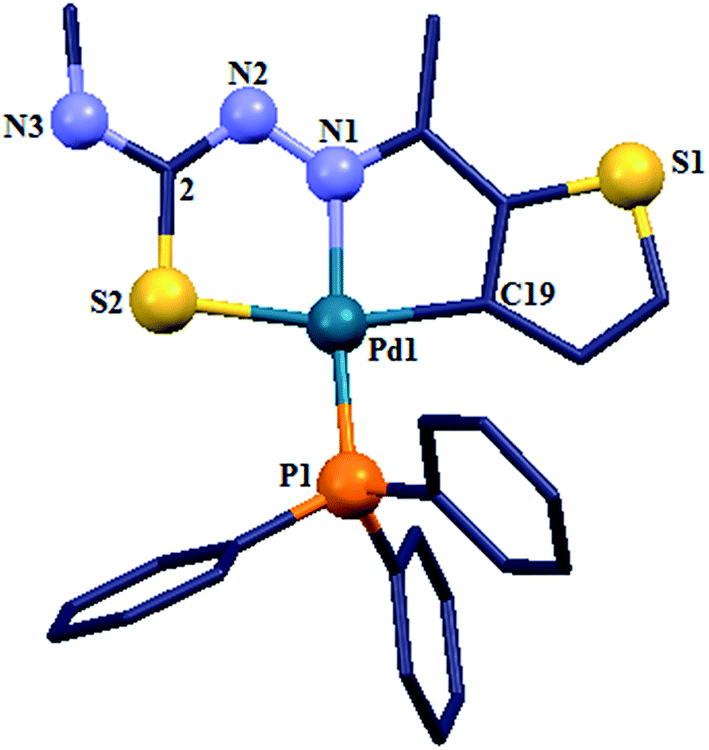
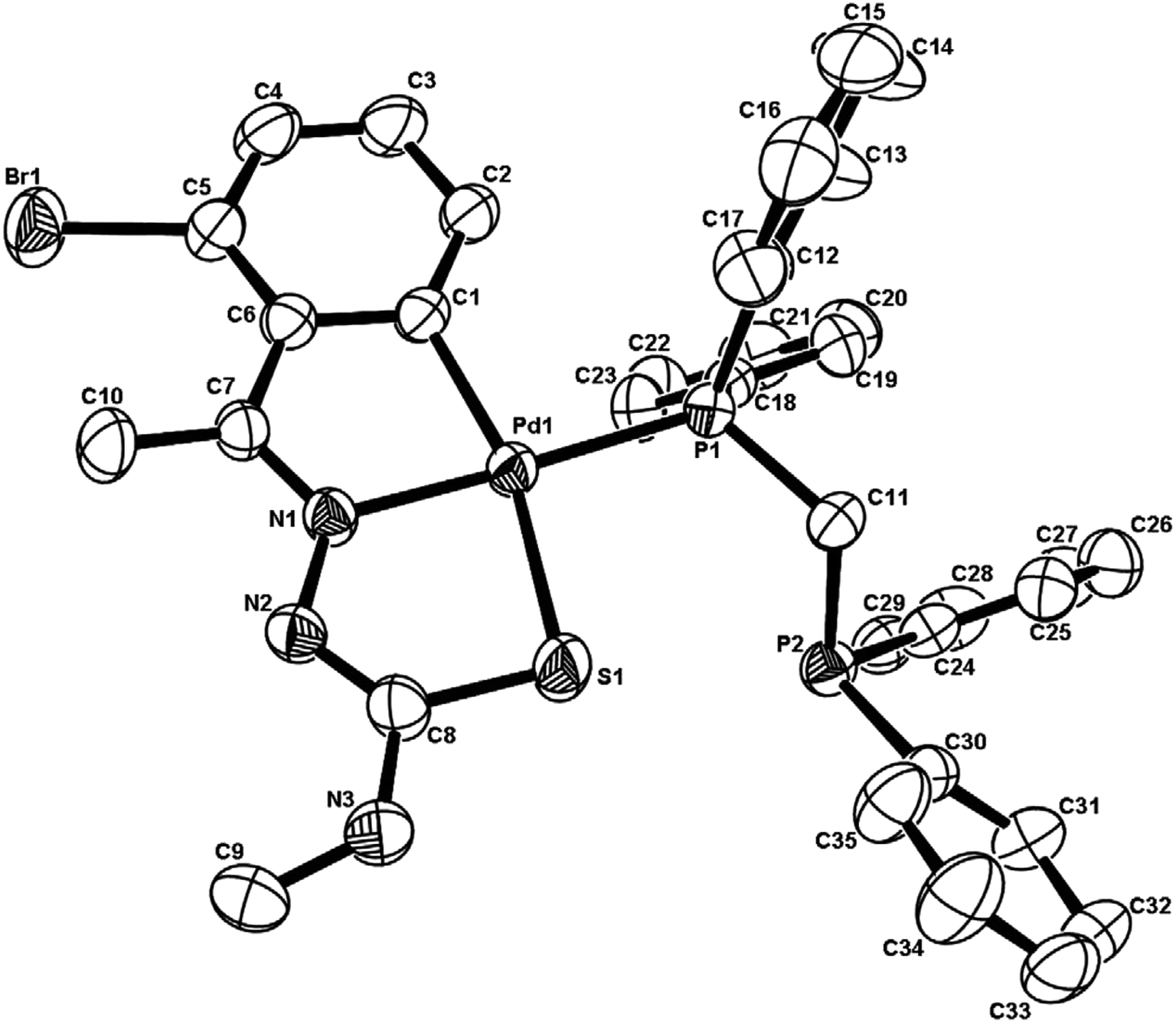
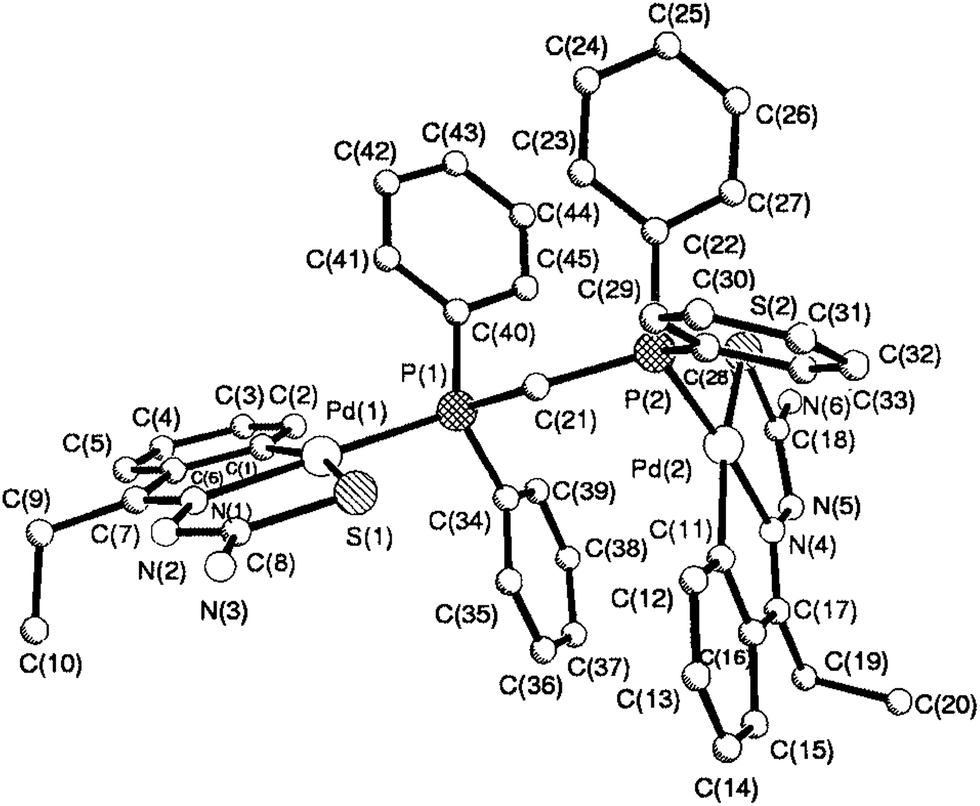
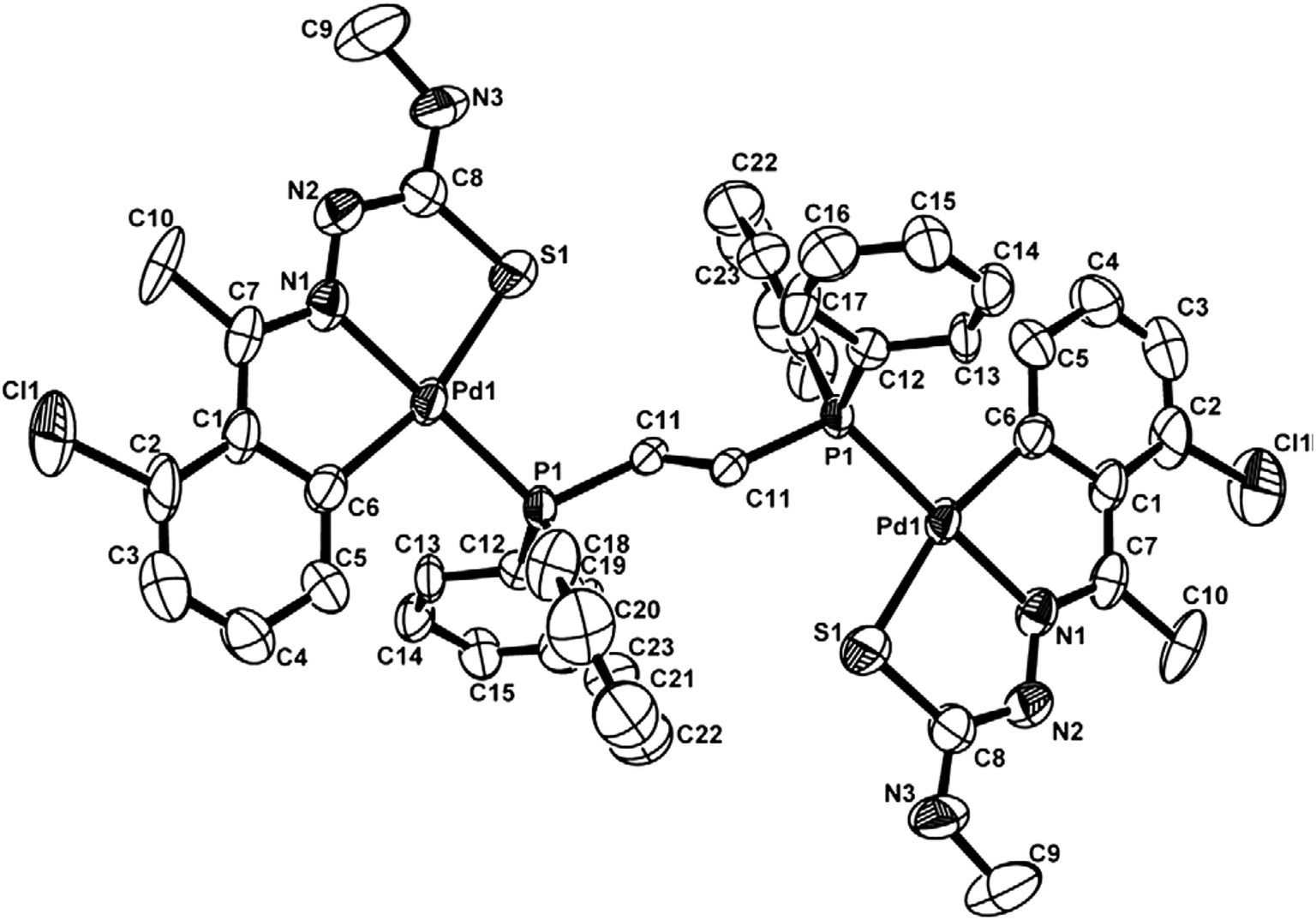
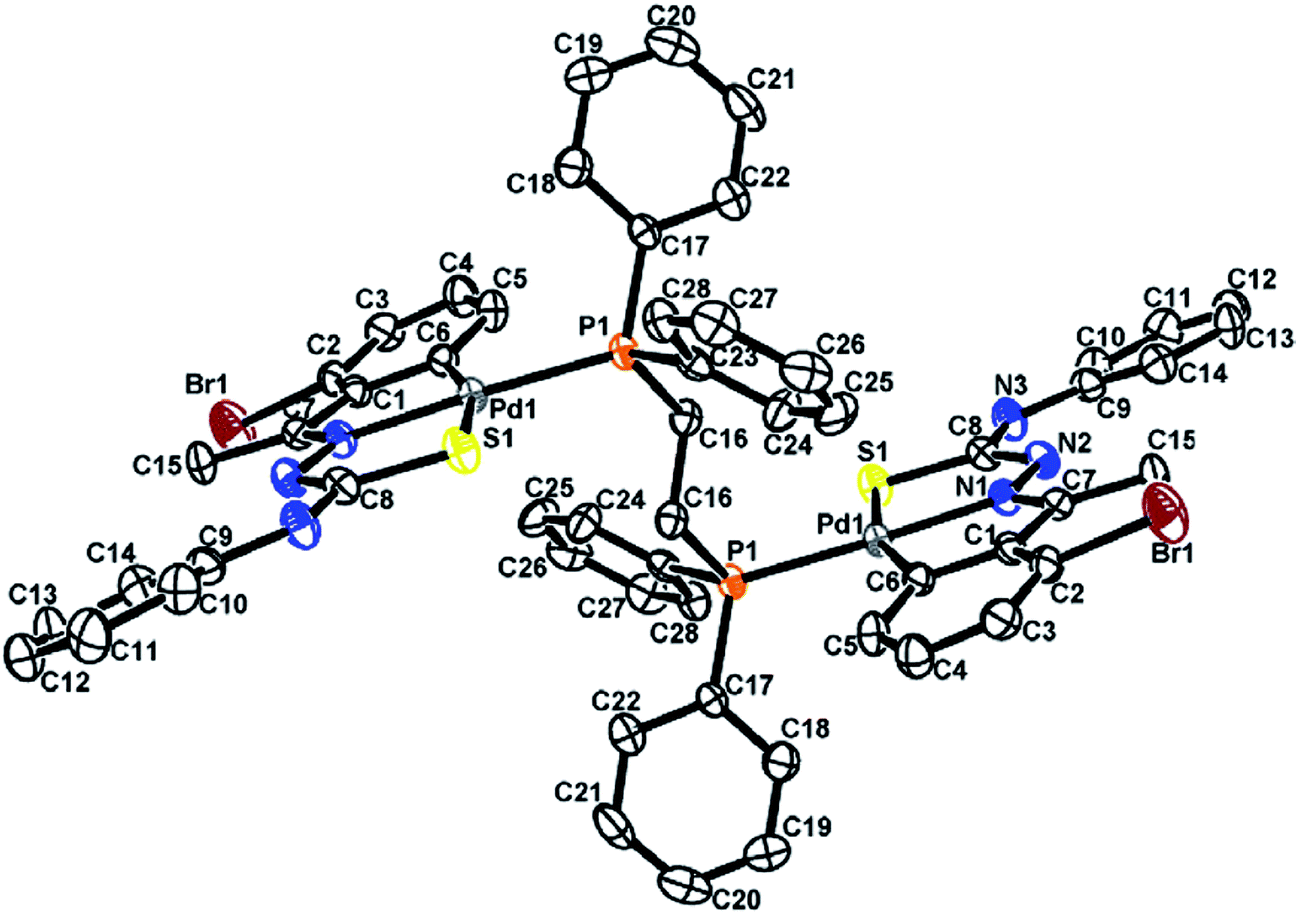
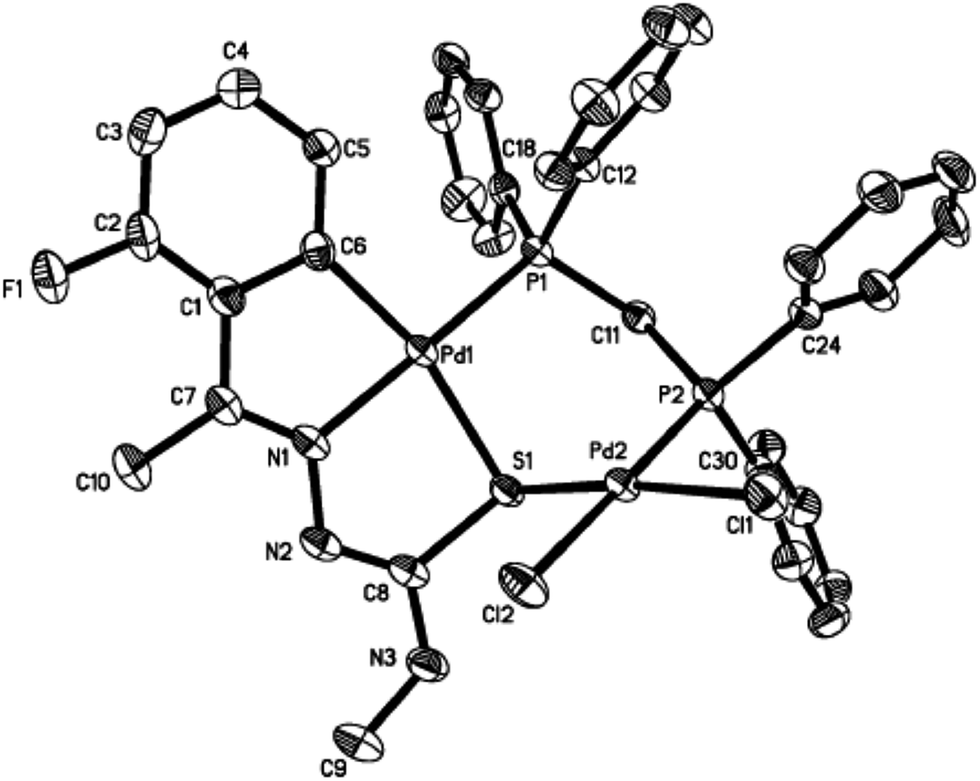
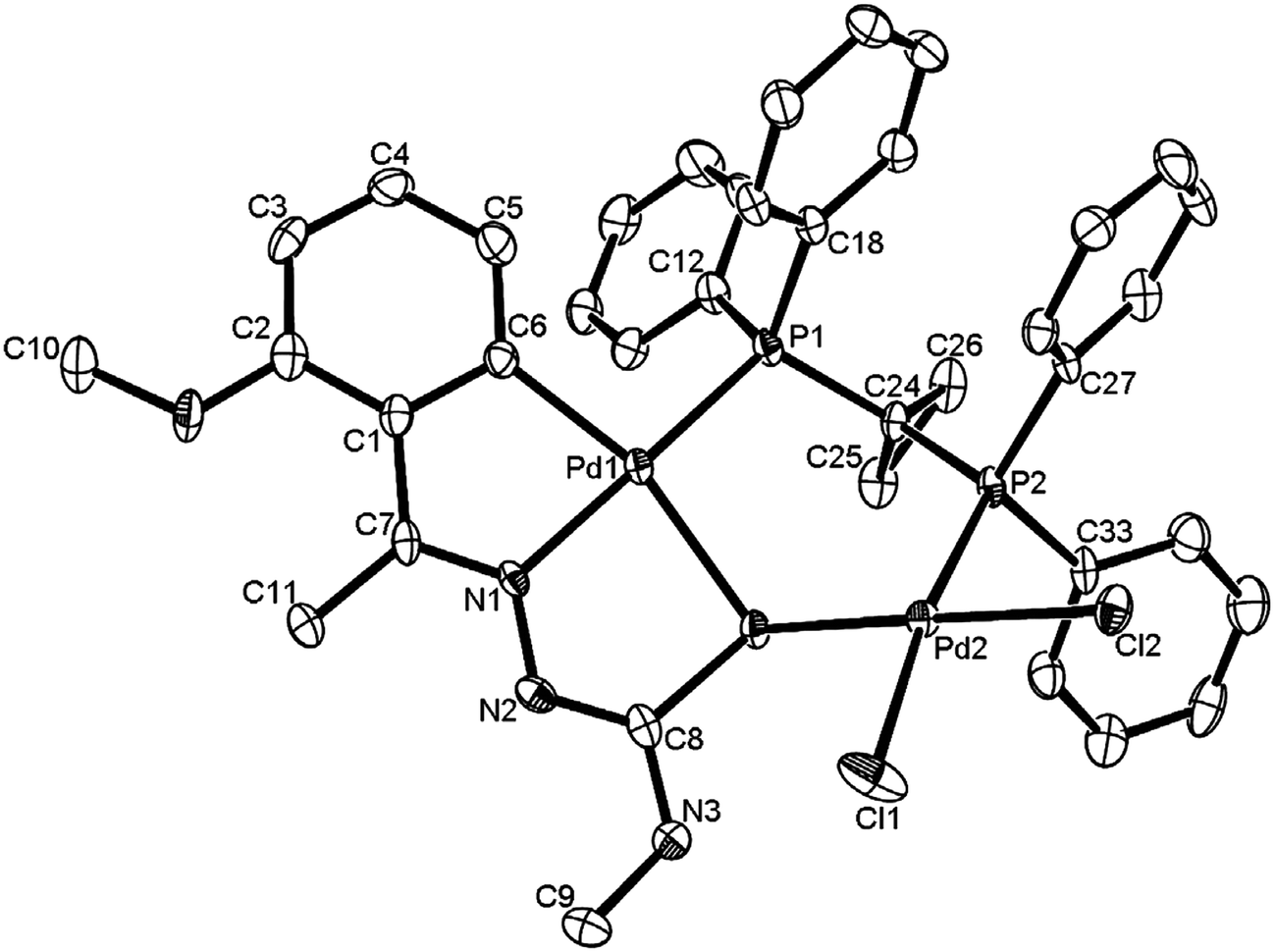
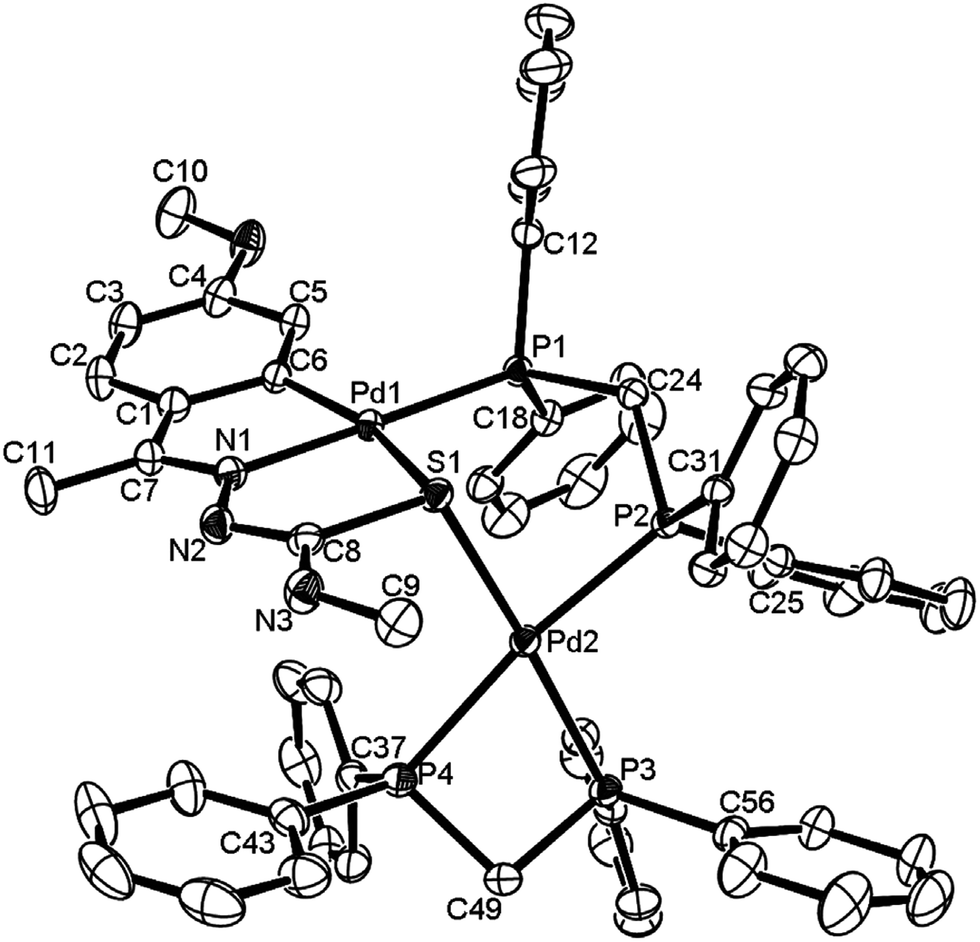
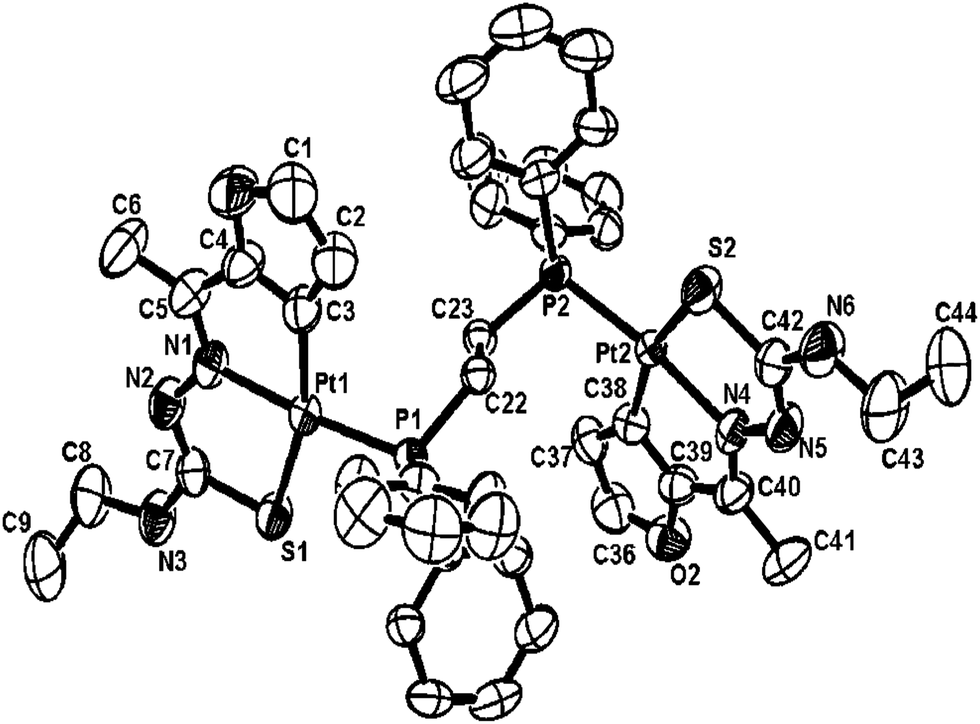
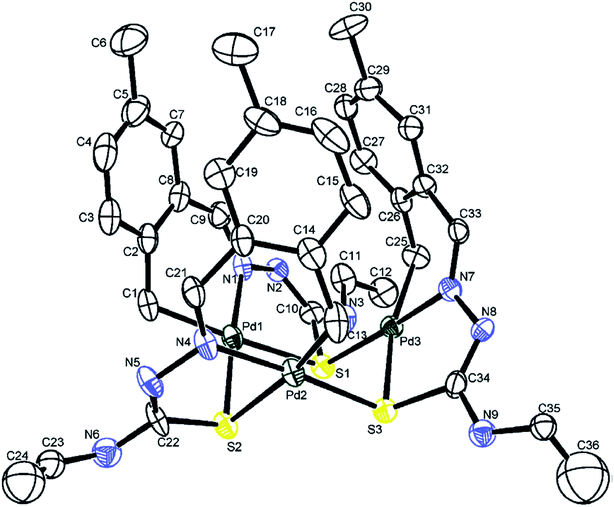
The co-ligands, dppm and dppen are bridging bidentate in dinuclear complexes, {Pd(C,N,S-L24)}2(μ-P,P-dppm) 44 (Fig. 10) and {Pd(C,N,S-L26)}2(μ-P,P-dppen) 75 (Fig. 11). Bis(tertiaryphosphines), dppe and dppp are only bridging bidentate in dinuclear complexes, {Pd(C,N,S-L31)}2(μ-P,P-dppe) 67 (Fig. 12) and {Pd(C,N,S-L28)}2(μ-P,P-dppp) 62 (Fig. 13). In another category of dinuclear complexes, [Pd2Cl2(C,N,μ-S-L32)(μ-P,P-dppm)] 97 (Fig. 14), [Pd2Cl2(C,N,μ-S-L46)(μ-P,P-bdcp)] 141 (Fig. 15) and [Pd2(C,N,μ-S-L41)(μ-P,P-dppm)(κ2-P,P-dppm)](ClO4)2 143 (Fig. 16), the co-ligands dppm and bdcp are bridging two Pd metal centers which are differently coordinated. One metal center has Pd(CNSP) (97, 141, 143) core and second metal center has Pd(PSCl2) (97, 141) or Pd(P3S) (143) core.
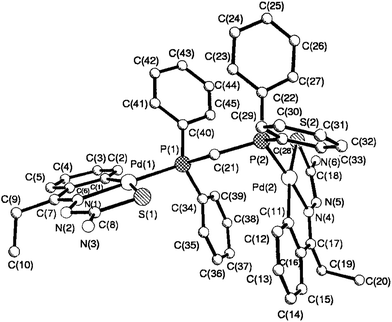 |
| | Fig. 10 Molecular structure of dinuclear{Pd(C,N,S-L24)}2(μ-P,P-dppm) 44 (L24: R2 = Et, R3 = H, R = H) [reproduced with permission from ref. 102. Copyright (1999) Royal Society of Chemistry]. | |
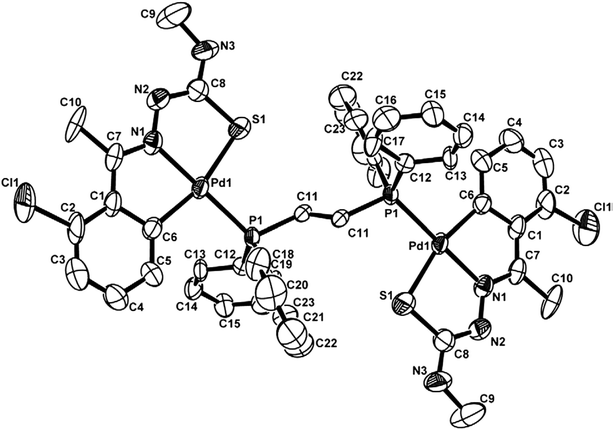 |
| | Fig. 11 Molecular structure of {Pd(C,N,S-L26)}2(μ-P,P-dppen) 75 (L26: R2 = Me, R3 = Me, R = Cl at 4-position of ring) [reprinted with permission from ref. 108 Copyright (2006) Elsevier]. | |
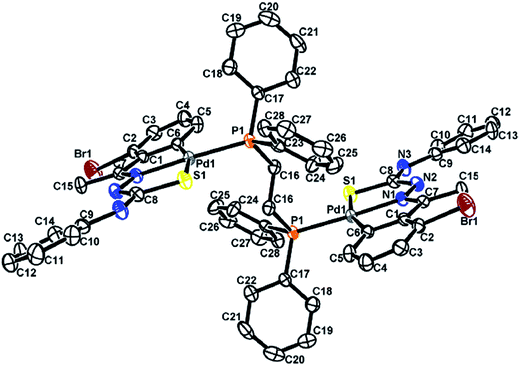 |
| | Fig. 12 Molecular structure of {Pd(C,N,S-L31)}2(μ-P,P-dppe) 67 (L31: R2 = Me, R3 = Ph, R = Br at 4-position of ring) [reprinted with permission from ref. 106. Copyright (2012) Elsevier]. | |
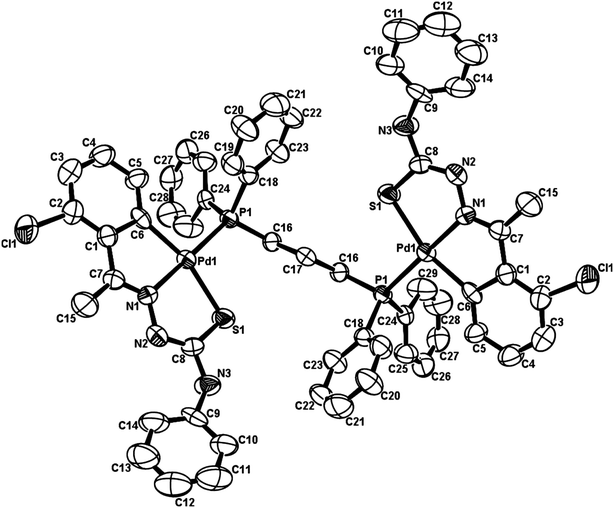 |
| | Fig. 13 Molecular structure of {Pd(C,N,S-L28)}2(μ-P,P-dppp) 62 (L28: R2 = Me, R3 = Ph, R = Cl at 4-position of ring) [reprinted with permission from ref. 105. Copyright (2006) Elsevier]. | |
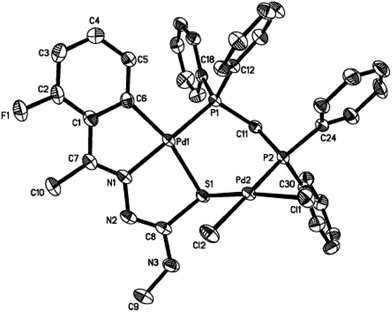 |
| | Fig. 14 Molecular structure of [Pd2Cl2(C,N,μ-S-L32) (μ-P,P-dppm)] 97 (L32: R2 = Me, R3 = Me, R = F at 4-position of ring) [reprinted with permission from ref. 104. Copyright (2003) American Chemical Society]. | |
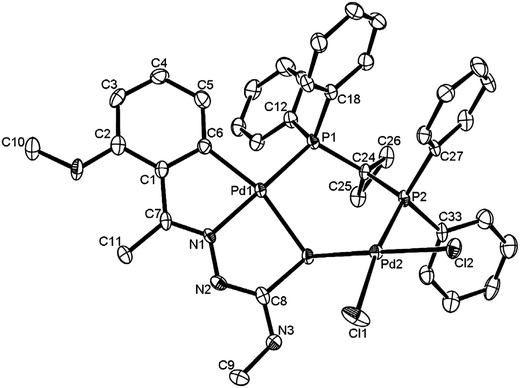 |
| | Fig. 15 Molecular structure of [Pd2Cl2(C,N,μ-S-L46) (μ-P,P-bdcp)] 141 (L46: R2 = Me, R3 = Me, R = MeO at 4-position of ring) [reprinted with permission from ref. 110. Copyright (2013) Elsevier]. | |
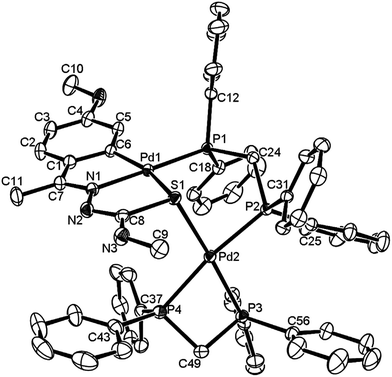 |
| | Fig. 16 Molecular structure of [Pd2(C,N,μ-S-L41)(μ-P,P-dppm)(κ2-P,P-dppm)](ClO4)2 143 (L41: R2 = Me, R3 = Me, R = MeO at 6-position of ring) (perchlorates not shown) [reprinted with permission from ref. 110. Copyright (2013) Elsevier]. | |
Platinum. There is one tetranuclear (182),96 one dinuclear (198)117 and two mononuclear (170, 187)115,116 platinum(II) compounds whose crystal structures have been determined. The coordination pattern of thio-ligands in tetranuclear PtII compound, {Pt4(C,N,μ-S-L4-2H)4} 182 (Fig. 17), mononuclear [Pt(C,N,S-L60-2H)(Ph3P)] 170 (Fig. 18) and [Pt(C,N,S-L65-2H)(Ph3P)] 187 (Fig. 19) as well as di-nuclear {Pt2(C,N,S-L70-2H)2}(dppe) 198 (Fig. 20) is similar to the corresponding PdII analogous compounds discussed in the preceding section.
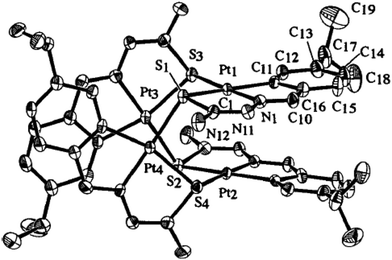 |
| | Fig. 17 Molecular structure of {Pt4(C,N,μ-S-L4-2H)4} 182 (L4: R2 = H, R3 = H; R = –CHMe2 at 6-position [reproduced with permission from ref. 96. Copyright (1998) American Chemical Society]. | |
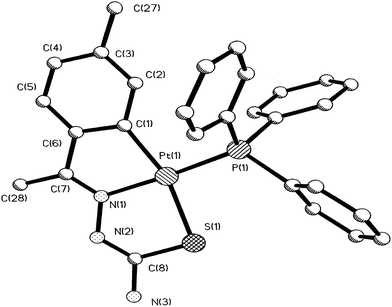 |
| | Fig. 18 Molecular structure of [Pt(C,N,S-L60-2H)(Ph3P)] 170 (L60: R2 = Me, R3 = H, R = Me at 6-position of ring) [reprinted with permission from ref. 115. Copyright (2000) Elsevier]. | |
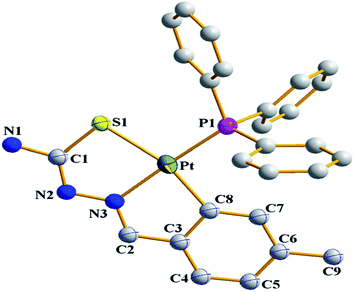 |
| | Fig. 19 Molecular structure of [Pt(C,N,S-L65-2H)(Ph3P)] 187 (L65: R2 = H, R3 = H, R = Me at 6-position of ring) [reprinted with permission from ref. 116. Copyright (2012) Elsevier]. | |
 |
| | Fig. 20 Molecular structure of {Pt2(C,N,S-L70-2H)2}(dppe) 198 (L70: R2 = Me, R3 = Et) [reproduced with permission from ref. 117 Copyright (2007) Wiley-VCH Verlag GmbH & Co. KGaA]. | |
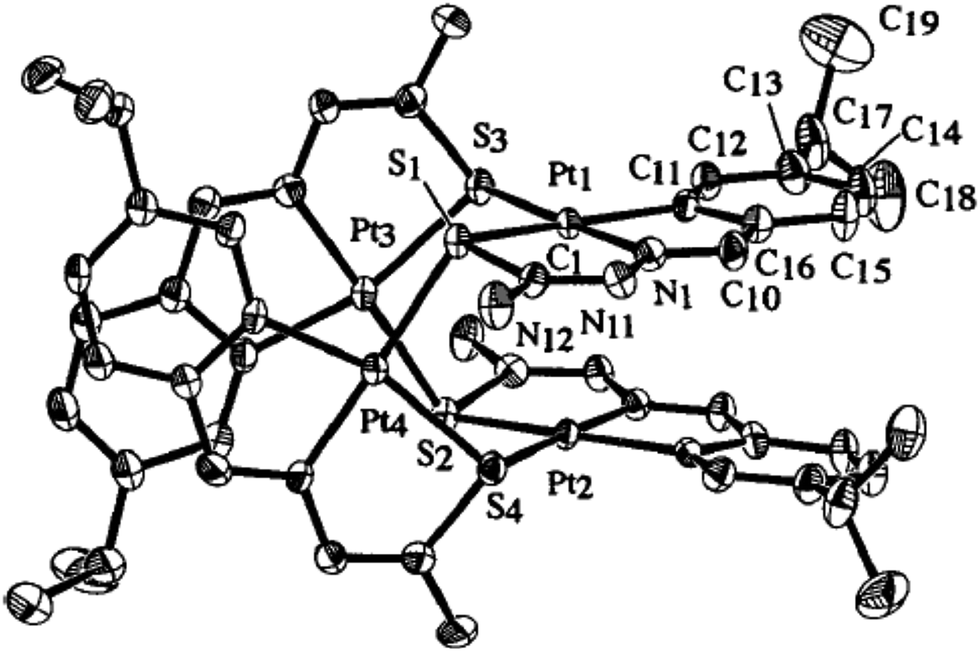
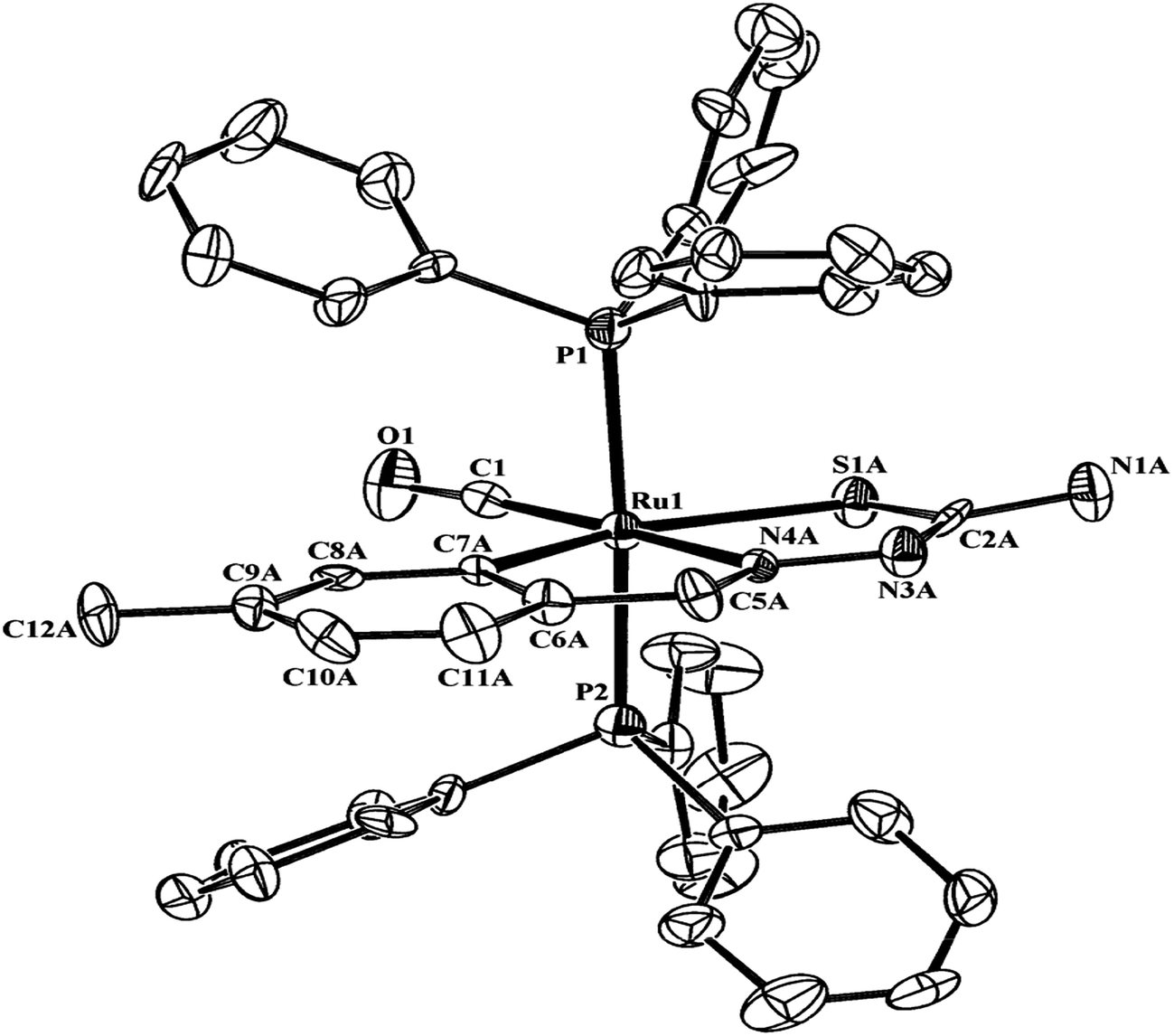
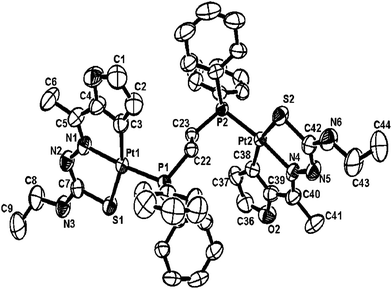
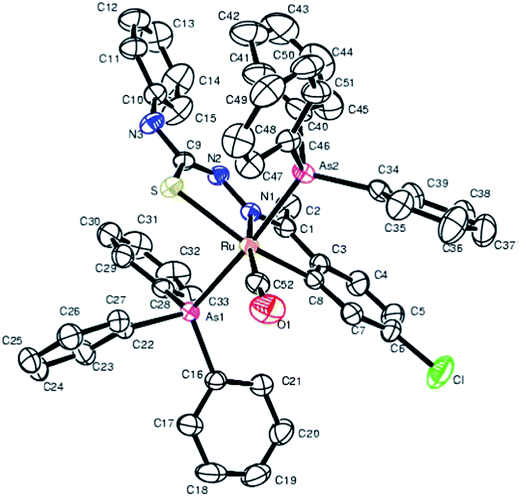
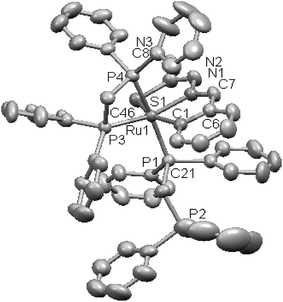
Ruthenium, rhodium and iridium. For ruthenium(II), molecular structures of five mononuclear (208, 220, 228, 231, 232)118,120–122 and two dinuclear (215, 230)119,121 compounds have been reported. Finally molecular structures of three mononuclear rhodium(III) (236, 238, 242)123 and two mononuclear iridium(III) (248, 250)124 have been determined. The co-ligands PPh3 and AsPh3 are trans in mononuclear complexes [Ru(C,N3,S-L72-2H)(CO)(PPh3)2] 208 (Fig. 21) and [Ru(C,N,S-L79-2H)(CO)(AsPh3)2] 220 (Fig. 22) with terdentate thio-ligands (C,N,S) coordinating in square plane with fourth site occupied by CO ligand. Thiophene based thio-ligand L88 (D type) as well as thio-ligand L89 (A type) coordinated as terdentate (C,N,S) to square planes of octahedral [Ru(C,N,S-L88-2H)(P,P-dppm)(P-dppm)] 231 (Fig. 23) and [Ru(C,N,S-L89-2H)(P,P-dppm)(P-dppm) 232 (Fig. 24) respectively. Here one dppm chelates RuII with one P donor atom occupying fourth site of square plane and second P donor atom binding to metal center along axial side. The sixth site along axial side is occupied by one P donor end of second dppm molecule leaving second donor end, –PPh2 pendant. Dinuclear RuII complex, [Ru2(C,N,μ-S-L74)2(PPh3)2(CO)2] 215 has one CO group and C,N,S-donor set of one thio-ligand coordinates the square plane and two such square planes are bridged through the coordinated sulfur and two PPh3 molecules coordinate along the terminal axial sides (Fig. 25). In mononuclear octahedral RhIII/IrIII compounds (236, 238, 242, 248 and 250),123,124 two PPh3 ligands in each case are trans along the axial side, while terdenate (C,N,S) thio-ligands occupy the square plane of the metal centers and fourth side in the plane is occupied by H− or Cl− donor groups (Fig. 26–30).
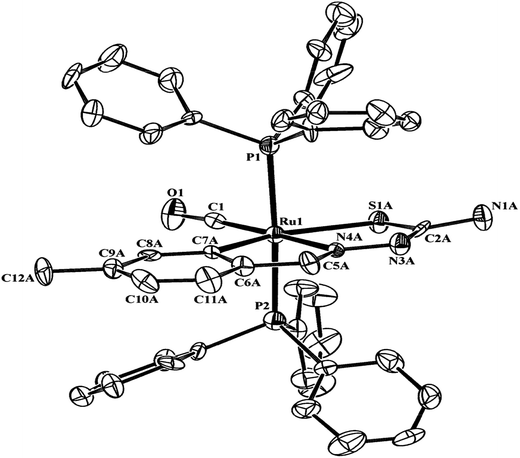 |
| | Fig. 21 Molecular structure of [Ru(C,N3,S-L72-2H)(CO)(PPh3)2] 208 (L72: R2 = H, R3 = H, R = Me at 6-position of ring) [reprinted with permission from ref. 118. Copyright (2011) Elsevier]. | |
 |
| | Fig. 22 Molecular structure of [Ru(C,N,S-L79-2H)(CO)(AsPh3)2] 220 (L79: R2, R3 = Me, Ph; R = Cl at 6-position) [reprinted with permission from ref. 120. Copyright 2009 Elsevier]. | |
 |
| | Fig. 23 Molecular structure [Ru(C,N,S-L88-2H)(P,P-dppm)(P-dppm)] 231 (L88: R2, R3 = H, H; see ligand type D above) [reprinted with permission from ref. 122. Copyright (2008) American Chemical Society]. | |
 |
| | Fig. 24 Molecular structure of [Ru(C,N,S-L89-2H)(P,P-dppm)(P-dppm) 232 (L89: R2, R3, R = H, H, H) [reprinted with permission from ref. 122. Copyright (2008) American Chemical Society]. | |
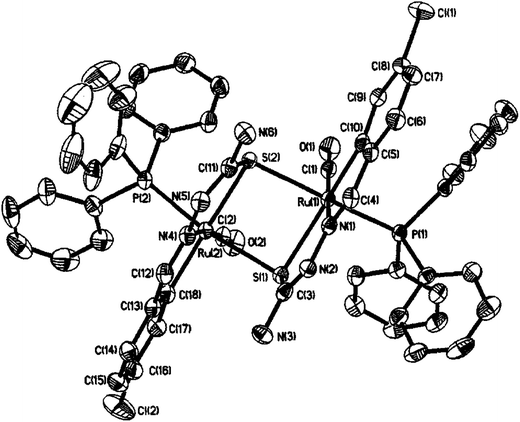 |
| | Fig. 25 Molecular structure of [Ru2(C,N,μ-S-L74-2H)2(PPh3)2(CO)2] 215 (L74: R2, R3 = H, H; R Cl at 6-position) [reproduced with permission from ref. 119. Copyright (2008) Wiley-VCH Verlag GmbH & Co. KGaA]. | |
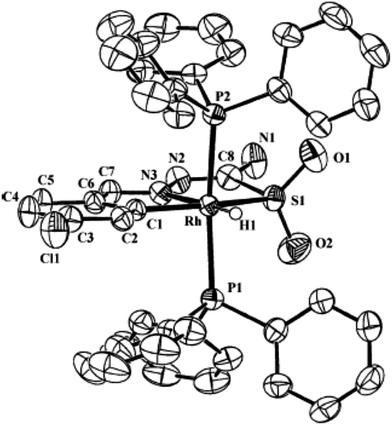 |
| | Fig. 26 Molecular structure of [Rh(C,N,S(O)2-L93-2H)(H)(PPh3)2] 236 (L93: R2, R3 = H, H; R = Cl at 6-position) [reprinted with permission from ref. 123. Copyright (2006) American Chemical Society]. | |
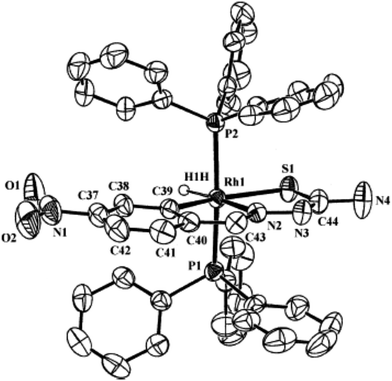 |
| | Fig. 27 Molecular structure of [Rh(C,N,S-L94-2H)(H)(PPh3)2] 238 (L94: R2, R3 = H, H; R = NO2 at 6-position) [reprinted with permission from ref. 123. Copyright (2006) American Chemical Society]. | |
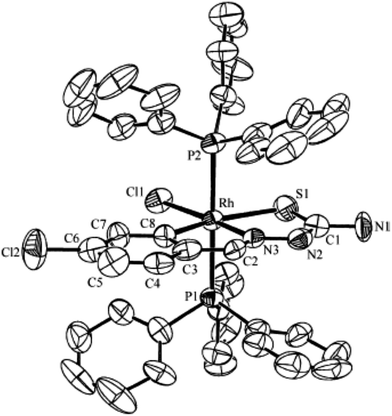 |
| | Fig. 28 Molecular structure of [Rh(C,N,S-L93-2H)(H)(PPh3)2] 242 (L93: R2, R3 = H, H; R = Cl at 6-position) [reprinted with permission from ref. 123. Copyright (2006) American Chemical Society]. | |
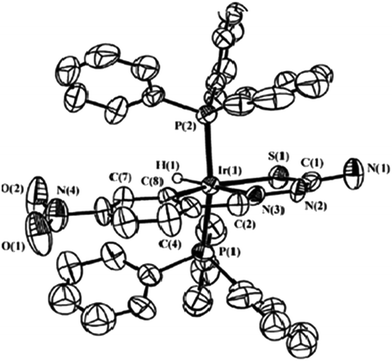 |
| | Fig. 29 Molecular structure of [IrH(C,N,S-L94-2H)(PPh3)2] 248 (L94: R2, R3 = H, H; R = NO2) at 6-position [reprinted with permission from ref. 124. Copyright 2010 Elsevier]. | |
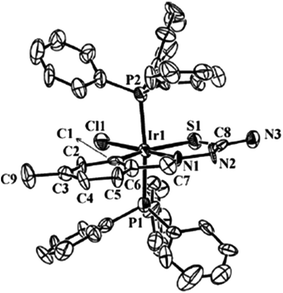 |
| | Fig. 30 Molecular structure of [Ir(C,N,S-L91-2H)(PPh3)2(Cl)] 250 (L91: R2, R3 = H, H; R = Me at 6-position) [reprinted with permission from ref. 124. Copyright 2010 Elsevier]. | |
The basic coordination pattern of thio-ligands around a metal center of tetranuclear PdII complexes (Table 1) belongs to E type core, except in complex 10 which has F type core and trinuclear complex 14 which has G type core (Chart 1). In E type core, metal forms 5-membered metallacyclic ring, and in F and G type cores, there are 6-membered rings. Due to the difference in nature of rings, the angles around a metal center are different. For example, in complex 27 (E core), the trans bond angles, N3–Pd–S (bridging) and C–Pd–S are 176.35(18) and 162.6(3)° respectively. These angles change to respective 173.23(12) and 176.29(14)° in 10 (F core) and 176.5(30) and 165.29(14)° in 14 (G core). Similarly, other angles C–Pd–N3 and N3–Pd–S are affected. The Pd–C, Pd–N and Pd–S bond distances differ to a small extent. The bond parameters of tetranuclear compounds 50, 53, 55, 100, 102 and 182 are similar to that of 27 (Table 1). The geometry around Pd/Pt metal centers in oligomers can be labeled as distorted square planar.
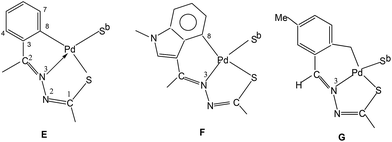 |
| | Chart 1 Basic coordination cores of complexes of different nuclearity; b refers to bridging sulfur. | |
It may be pertinent to note that compounds 9–13 have F type coordination pattern for thio-ligands, only 14 belongs to G type core. All other compounds 1–8, 15–50, 51–100, 101–150, 151–200, 201–253 described in Section 3.1 (Table 1 structurally characterized) have E type coordination pattern (Sb-bridging sulfur is replaced by P donor in phosphine complexes). A brief commentary on bond parameters of mono- and di-nuclear Pd/Pt complexes with phosphines as co-ligands is provided here. The coordination patterns of phosphines in mono- and di-nuclear Pd/Pt complexes are given in Chart 2. In case of PdII complexes, all the five patterns (H–L, Chart 2) have been observed, while in case of PtII only two patterns (H, J) were observed. The trans bond angles, N3–M–P and C–M–S fall in the ranges, 176–179 and 162–165° respectively while C–M–N3 and S–M–N3 angles lie in the range, 80–83°. Various M–C, M–N, M–S and M–P bond distances are normal and are only marginally different. The C–S bond distances suggest the presence of partial double bond character. Ruthenium(II) complexes have shown four types (M, N, O, P) of coordination patterns as shown in Chart 3. Rhodium(III) and iridium(III) complexes belong to only M type coordination pattern. The trans bond angles suggest distorted octahedral geometries of complexes of these metals (see Table 1).
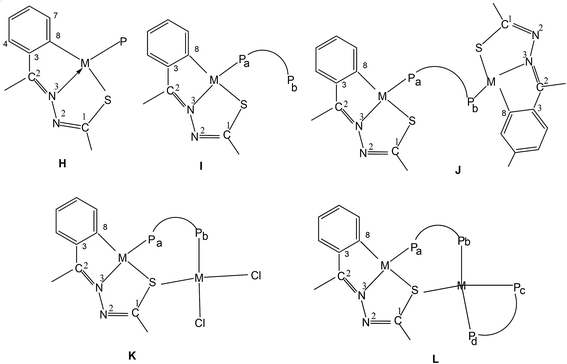 |
| | Chart 2 Different coordination patterns of phospines in Pd/Pt complexes. | |
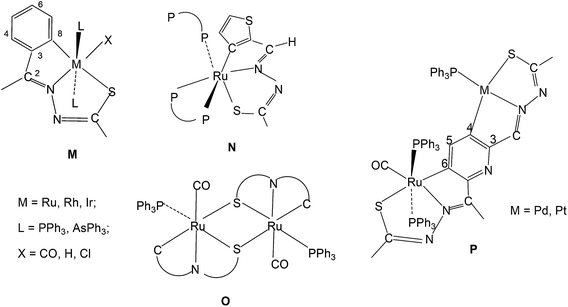 |
| | Chart 3 Coordination patterns of Ru, Rh and Ir complexes. | |
3.3 Electronic absorption spectroscopy, fluorescence and cyclic voltammetry
The reported electronic absorption spectral data of metal complexes are placed in Table 2.101,113,118–121,123,124 The assignments have been supported by DFT calculations in several cases using standard model procedures. In PdII compounds, the transitions occur from π → π* (Region I), n → π* orbitals in Regions II and III. In case of ruthenium compounds, π → π* and n → π* transitions are assigned in high energy regions I and II and in low energy visible region (III), the MLCT transitions (metal to ligand charge transfer) involve movement of metal t2g electron to π* orbitals on –CN-(azomethine imine) moiety.118–121 The behavior of Rh/Ir compounds is similar to that of Ru compounds.123,124 Mononuclear and dinuclear complexes did not differ much in transitions. Only one report deals with fluorescence spectroscopy. Complexes 217–227 (Ru) showed weak emission in the region, 590–596 nm, corresponding to excitation wave length of 450 nm.120
Table 2 Electronic absorption spectral data of complexesa
| Compounds |
Region III nm |
Region II nm |
Region I nm |
Ref. |
| M-mononuclear; D-dinuclear. |
| Pd: 17–21 (M) |
452 to 494 L → L |
326–364 L → L |
272–262 L → L |
101 |
| Pd: 157–158 (M) |
429–447 L → L |
336–368 L → L |
266–272 L → L |
113 |
| Ru: 207–211 (M) |
462–547 L → L |
342–400; 300–338 L → L |
269–272 L → L |
118 |
| Ru: 212–216 (D) |
436–562 MLCT |
302–396 L → L |
270–352 L → L |
119 |
| Ru: 217–227 (M) |
462–580 MLCT |
307–398 L → L |
242–250 L → L |
120 |
| Ru: 228 (M) Ru–Pd/Pt: 229–230 (D) |
492, 480–488 MLCT |
334–400 L → L |
276–304 L → L |
121 |
| Rh: 233–243 (M) |
400–510 MLCT |
290–360 L → L |
234–286 L → L |
123 |
| Ir: 244–253 (M) |
436–510 MLCT |
— |
232–382 L → L |
124 |
Only complexes of Ru, Rh and Ir are studied for their cyclic voltammetric studies. The CV study of RuII complexes 207 to 211 in CH2Cl2–CH3CN mixture (tetrabutylammonium hexafluro phosphate, TBAP, as supporting electrolyte) showed two oxidative responses in the region 0.46 to 1.33 V and one reductive response in the region, −1.00 to −1.41 V relative to SCE. These are attributed to the oxidation (former) and reduction (latter) of the coordinated ligand.118 The CV of dinuclear complexes 212 to 216 in CH2Cl2–CH3CN mixture using tetrabutylammonium perchlorate as supporting electrolyte have shown reversible RuII–III redox behavior {E1/2 – 0.48 to 0.70 V; ΔEp (Epa − Epc) = 70 to 80 mV}. Additionally dinuclear complexes have shown an irreversible oxidative response in the range, 1.09 to 1.47 V and this is assigned to RuIII–RuIV oxidation.119 Mononuclear RuII complexes 217 to 227 exhibit a quasi reversible one electron reduction RuII/RuI) in the range −0.83 to −0.86 V (SCE, TBAP) and it is noted that the formal potential of all the couples appear to correlate linearly with the Hammett constant of the p`phenyl of the acetophenone thiosemicarbazone ligands.120
The CV study of RhIII complexes 233 to 243 showed two oxidative responses in the regions, 0.64 to 1.07 V (reversible in some cases), 0.93 to 1.73 V and one reductive response in the region −1.06 to −1.32 V relative to SCE. The first oxidative response is assigned to RhIII–RhIV oxidation, second irreversible oxidative response to the coordinated thiosemicarbazone and finally the reductive response is attributed to reduction of the coordinated thiosemicarbazone.123 Iridium(III) complexes 244 to 253 has also shown two oxidative responses in the regions, 0.47 to 0.92 V (reversible), 1.07 to 1.36 V and one reductive response in the region −0.74 to −1.47 V relative to SCE. The first oxidative response is assigned to IrIII–IrIV oxidation, second irreversible oxidative response to the coordinated thiosemicarbazone and finally the reductive response is attributed to reduction of the coordinated thiosemicarbazone.124
3.4 NMR spectroscopy
The ortho-metallated compounds have also been studied using 1H, 13C and 31P NMR spectroscopy (see structure E for numbering scheme of a thio-ligand in general). The 1H NMR spectral data are given only for representative examples and are placed in Table 3. The reported 13C and 31P NMR data are given for significant or diagnostic signals for nearly all the compounds which are listed in Table 4 and 5 respectively. The 1H NMR spectral signals are first commented briefly here. For example, tetra-nuclear oligomer 1 (ref. 95) showed –C2–H, –N1–H, –N1–Me and –OMe (R-ring) proton signals at δ = 6.78(s), 5.1(br), 2.3(d) and 3.87(s) ppm respectively which were mostly up-field relative to the free ligand L1 in this case. The –N2–H proton as expected disappeared in oligomer 1 which was at 9.66 ppm in the free ligand (Table 3). Several selected complexes listed in Table 3 are shown to demonstrate this trend and is true of all other complexes reported in literature. The –NH2 protons in the free ligands usually appear as a pair or a broad single signals, and generally as a single signal in complexes. Metal-hydride signals of Rh/Ir complexes appeared as multiplets (coupling from Rh/P nuclei, I = 1/2 in each case) in the high field region (Table 3).
Table 3 1H NMR data of representative complexes (δ in ppm)
|
| Compd. no. |
–C2–H |
–N2–H |
–N1–H |
–N1–R |
R-ringa |
(Ref.) |
| Refers to the substituent of ring at C2 carbon. Metallated Me group of ring. n.a. – not applicable, n.o. – not observed. Metal hydride, Rh–H; and Ir–H; dt = doublet of triplet; td = triplet of doublet. Obscured by PPh3. |
| L1 |
7.80 (s) |
9.66 (s) |
7.56 br |
3.26 (d, Me) |
3.84 (s, OMe) |
95 |
| 1 |
6.78 (s) |
— |
5.1 (br) |
2.3 (d, Me) |
3.87 (s, OMe) |
95 |
| L9 |
8.30 (s) |
11.48 (s) |
8.24, 7.61 (br) |
— |
2.68 (s, OMe) |
98 |
| 12 |
8.79 (d) JH–P=2.7 |
— |
n.o.c |
n.o.c |
2.66 (s, OMe) |
98 |
| L11 |
8.23 (s) |
10.31 (s) |
7.42 (s) |
1.31 (m), 3.76 (m) (Et) |
2.34, 2.40 (2 s, Me) |
99 |
| 14 |
7.91 (s) |
— |
4.99 (m) |
1.29 (m), 3.47 (m) (Et) |
2.23 s (Me), 3.10 (s, CH2)b |
99 |
| L12 |
7.84(s) |
3.74 s (N2–Me) |
8.22 s, 8.27 s |
n.a.c |
3.78 s (MeO) |
100 |
| 15 |
8.14 s |
3.46 (N2–Me) |
n.o.c |
n.a.c |
3.72 s (MeO) |
100 |
| L13 |
7.85 (s) |
3.76 s (N2–Me) |
8.31 s, 8.38 s |
n.a.c |
3.79 (MeO) |
100 |
| 16 |
8.18 s |
3.47 (N2–Me) |
9.47 s, 8.74 s |
n.a.c |
3.68 s (MeO) |
100 |
| 17 |
8.18 s |
— |
4.79 s (NH2) |
n.a.c |
3.84 s (MeO) |
101 |
| L63 |
2.28 s (C2–Me) |
8.70 br |
7.60 br |
3.25d (Me) |
2.38 s (Me) |
115 |
| 168 |
1.72 s (C2–Me) |
— |
5.20 br |
3.06d (Me) |
2.36 s (Me) |
115 |
| 173 |
1.75 s (C2–Me) |
— |
4.7 br |
2.96d (Me) |
2.44 s (Me) |
115 |
| 175 |
2.31 s |
— |
5.20 br |
n.a.c |
0.89 t (Me); 3.70 t (CH2O–) |
115 |
| 208 |
6.96 s |
— |
5.34 s |
n.a.c |
1.85 s(Me) |
118 |
| 213 |
7.78 s |
— |
4.04 s |
n.a.c |
1.94 s(Me) |
119 |
| 234 |
6.70 s |
–; −11.5 td |
4.69 s |
n.a.c |
1.91 s(Me) |
123 |
| 240 |
6.93 s |
— |
4.39 s |
n.a.c |
1.90 s(Me) |
123 |
| 245 |
7.29–7.66e |
–, −19.33 dtd −15.27 dtd |
4.49 s |
n.a.c |
1.99 s(Me) |
124 |
| 250 |
6.90 s |
— |
4.60 s |
n.a.c |
2.00 s(Me) |
124 |
Table 4 The 13C NMR data of complexes (δ in ppm)
| 13C NMRa |
| Compd. no. |
δ(C8) |
δ(C2) |
δ(C1(S)) |
δ(C3) |
Solvent |
(Ref.) |
| Reference TMS. |
| L1 |
129.2 |
142.0 |
177.9 |
127.2 |
CDCl3 |
95 |
| 1 |
148.0 |
156.2 |
167.9 |
132.1 |
CDCl3 |
95 |
| 2 |
141.9 |
158.8 |
168.2 |
132.6 |
CDCl3 |
95 |
| 4 |
165.7 |
158.7 |
167.8 |
147.41 |
dmso-d6 |
96 |
| 5 |
164.9 |
160.0 |
167.1 |
146.4 |
CDCl3 |
97 |
| 6 |
166.4 |
158.6 |
169.1 |
146.9 |
CDCl3 |
97 |
| 7 |
168.5 |
158.7 |
171.1 |
145.6 |
CDCl3 |
97 |
| 8 |
166.7 |
158.3 |
168.4 |
147.6 |
CDCl3 |
97 |
| 15 |
160.1 |
157.6 |
189.1 |
139.6 |
CDCl3 |
100 |
| 16 |
160.1 |
157.8 |
174.6 |
139.6 |
CDCl3 |
100 |
| 22 |
147.5 |
166.8 |
167.3 |
165.1 |
dmso-d6 |
102 |
| 23 |
147.2 |
165.9 |
169.6 |
165.1 |
CDCl3 |
100 |
| 24 |
147.5 |
165.8 |
169.1 |
165.0 |
CDCl3 |
100 |
| 25 |
147.0 |
166.5 |
172.1 |
161.0 |
CDCl3 |
100 |
| 27 |
148.5 |
167.2 |
172.0 |
165.5 |
dmso-d6 |
102 |
| 28 |
142.2 |
159.5 |
168.0 |
132.0 |
CDCl3 |
95 |
| 29 |
151.1 |
166.2 |
174.1 |
158.5 |
dmso-d6 |
102 |
| 33 |
149.0 |
165.6 |
175.2 |
163.7 |
dmso-d6 |
102 |
| 123 |
165.1 |
166.1 |
167.7 |
150.7 |
dmso-d6 |
109 |
| 124 |
165.5 |
168.1 |
170.8 |
149.4 |
dmso-d6 |
109 |
| 127 |
152.7 |
163.6 |
176.6 |
136.1 |
dmso-d6 |
109 |
| 128 |
164.1 |
168.2 |
176.9 |
151.5 |
dmso-d6 |
109 |
| 129 |
152.7 |
163.8 |
178.6 |
136.1 |
dmso-d6 |
109 |
| 130 |
163.3 |
164.0 |
176.6 |
152.6 |
dmso-d6 |
109 |
| 131 |
153.2 |
163.9 |
165.1 |
136.8 |
dmso-d6 |
109 |
| 132 |
151.4 |
168.3 |
176.8 |
136.2 |
dmso-d6 |
109 |
| 133 |
163.7 |
168.4 |
176.6 |
151.3 |
dmso-d6 |
109 |
| 134 |
164.7 |
169.1 |
177.4 |
152.1 |
dmso-d6 |
109 |
| 182 |
154.2 |
162.1 |
165.0 |
145.1 |
dmso-d6 |
96 |
| 183 |
154.0 |
162.0 |
165.0 |
145.6 |
CDCl3 |
97 |
| 184 |
158.9 |
160.8 |
167.3 |
146.2 |
CDCl3 |
97 |
| 185 |
156.9 |
161.2 |
Not obsd. |
146.7 |
CDCl3 |
97 |
Table 5 The 31P NMR data of complexes (δ in ppm; J in Hz)abc
| δP |
δP |
(Ref.) |
| 85% H3PO4. (MeO)3P reference markers. {M-mononuclear; D-dinuclear, M-P, mononuclear with pendant PPh2 group; M-C, mononuclear with chelating dppm donors facing different donors bonded to metal; D-UM, dinuclear with phosphines bridging unequal metals; D-UUM-dinuclear with unsymmetrical phosphines bonded to unequal metals; M-UP, mononuclear with unequal phosphines}. |
| 12D |
70.4 |
dmso-d6 |
13D |
70.3 |
dmso-d6 |
98 |
| 30M |
36.7 |
CDCl3 |
31M |
36.8 |
CDCl3 |
100 |
| 32M |
36.7 |
CDCl3 |
35M |
38.4 |
CDCl3 |
100 |
| 36M |
38.5 |
CDCl3 |
37M |
38.6 |
CDCl3 |
100 |
| 38M |
38.3 |
CDCl3 |
39D |
34.4 |
CDCl3 |
102 |
| 40D |
27.0 |
CDCl3 |
41D |
25.4 |
CDCl3 |
102 |
| 42D |
34.2 |
dmso-d6 |
43D |
27.4 |
CDCl3 |
102 |
| 44D |
25.4 |
CDCl3 |
|
|
CDCl3 |
102 |
| 45M-P |
27.6 (d, Pa; 2JPP = 71.0), −23.7 (d, Pb; 2JPP = 71.0) |
CDCl3 |
102 |
| 46M-P |
22.3 (d, Pa; 2JPP = 71.0), −30.2 (d,Pa; 2JPP = 71.0) |
CDCl3 |
102 |
| 47M-P |
42.7 (d, Pa; 2JPP = 65.7.0), −16.4 (d, Pa; 2JPP = 65.7) |
CDCl3 |
102 |
| 48M-P |
27.2 (d, Pa; 2JPP = 73.0), −24.0 (d, Pb; 2JPP = 73.0) |
CDCl3 |
102 |
| 58D |
23.7 |
CDCl3 |
59D |
22.9 |
CDCl3 |
103 |
| 60D |
31.5 |
CDCl3 |
61D |
32.5 |
CDCl3 |
105 |
| 62D |
26.8 |
CDCl3 |
63D |
28.7 |
CDCl3 |
105 |
| 64D |
23.6 |
CDCl3 |
65D |
23.7 |
CDCl3 |
103 |
| 66D |
23.7 |
CDCl3 |
67D |
32.5 |
CDCl3 |
106 |
| 68D |
26.8 |
CDCl3 |
69D |
28.7 |
CDCl3 |
106 |
| 70D |
32.5 |
CDCl3 |
|
|
|
106 |
| 71D |
23.8 |
CDCl3 |
72D |
31.8 |
CDCl3 |
108 |
| 73D |
32.0 |
CDCl3 |
74D |
31.4 |
CDCl3 |
108 |
| 75D |
32.4 |
CDCl3 |
76D |
32.4 |
CDCl3 |
108 |
| 77D |
26.9 |
CDCl3 |
78D |
26.9 |
CDCl3 |
108 |
| 79D |
31.6 |
CDCl3 |
80D |
32.3 |
CDCl3 |
107 |
| 81D |
26.9 |
CDCl3 |
82D |
28.7 |
CDCl3 |
107 |
| 83M-P |
24.1 (d, Pa; 2JPP = 79.8), −26.7 (d, Pb; 2JPP = 79.8) |
CDCl3 |
103 |
| 84M-P |
23.7 (d, Pa; 2JPP = 79.8), −26.7 (d, Pb; 2JPP = 79.8) |
CDCl3 |
103 |
| 85M-P |
23.9 (d, Pa; 2JPP = 79.8), −26.8 (d, Pb; 2JPP = 79.8) |
CDCl3 |
103 |
| 86M-P |
23.6 (d,; Pa 2JPP = 75.1), −26.7 (d, Pb; 2JPP = 75.1) |
CDCl3 |
103 |
| 87M-P |
23.7 (d, Pa; 2JPP = 75.1), −26.7 (d, Pb; 2JPP = 75.1) |
CDCl3 |
106 |
| 88M-P |
24.4 (d, Pa; 2JPP = 75.1), −26.3 (d, Pb; 2JPP = 75.1) |
CDCl3 |
108 |
| 89M-P |
24.4 (d, Pa; 2JPP = 75.1), −26.3 (d, Pb; 2JPP = 75.1) |
CDCl3 |
108 |
| 90M-P |
22.3 (d, Pa; 2JPP = 75.3), −25.2 (d, Pb; 2JPP = 75.3) |
CDCl3 |
108 |
| 91M-C |
24.4 (d, Pa; 2JPP = 13.1); 22.8 (d, Pb; 2JPP = 13.3) |
CDCl3 |
103 |
| 92M-C |
24.3 (d, Pa; 2JPP = 13.1); 22.7 (d, Pb; 2JPP = 13.1) |
CDCl3 |
103 |
| 93M-C |
24.1 (d, Pa; 2JPP = 13.1), 22.7 (d, Pb; 2JPP = 13.1) |
CDCl3 |
103 |
| 94M-C |
24.2 (d, Pa; 2JPP = 13.7); 22.8 (d, Pb; 2JPP = 14.5) |
CDCl3 |
103 |
| 95D-UM |
22.5 (d, Pa; 2JPP = 26.0); 16.7 (d, Pb) |
CDCl3 |
104 |
| 96D-UM |
22.9 (d, Pa; 2JPP = 26.0); 16.8 (d, Pb) |
CDCl3 |
104 |
| 97D-UM |
22.7 (d, Pa; 2JPP = 26.7); 16.8 (d, Pb) |
CDCl3 |
104 |
| 98D-UM |
22.9 (d, Pa; 2JPP = 28.5); 16.8 (d, Pb) |
CDCl3 |
104 |
| 103D |
32.1 |
CDCl3 |
104D |
31.9 |
CDCl3 |
105 |
| 105D |
27.1 |
CDCl3 |
106D |
28.7 |
CDCl3 |
105 |
| 107D |
23.6 |
CDCl3 |
108D |
32.0 |
CDCl3 |
106 |
| 109D |
27.1 |
CDCl3 |
110D |
8.7 |
CDCl3 |
106 |
| 111D |
32.7 |
CDCl3 |
|
|
|
106 |
| 112D |
31.5 |
CDCl3 |
113D |
32.5 |
CDCl3 |
105 |
| 114D |
26.8 |
CDCl3 |
115D |
28.7 |
CDCl3 |
105 |
| 116D |
24.2 |
CDCl3 |
117D |
32.4 |
CDCl3 |
106 |
| 118D |
27.3 |
CDCl3 |
119D |
29.2 |
CDCl3 |
106 |
| 120D |
32.9 |
CDCl3 |
|
|
|
106 |
| 121M-P |
24.4 (d, Pa; 2JPP = 75.1), −26.5 (d, Pb; 2JPP = 75.1) |
CDCl3 |
106 |
| 122M-P |
25.0 (d, Pa; 2JPP = 75.1), −26.4 (d, Pb; 2JPP = 75.1) |
CDCl3 |
106 |
| 127M |
38.8 |
dmso-d6 |
128M |
38.6 |
dmso-d6 |
109 |
| 129D |
29.2 |
dmso-d6 |
130D |
34.2 |
dmso-d6 |
109 |
| 131D |
39.0 |
dmso-d6 |
132D |
29.1 |
dmso-d6 |
109 |
| 133D |
33.7 |
dmso-d6 |
134D |
38.8 |
dmso-d6 |
109 |
| 135D-UM |
23.2 (d, Pa; 2JPP = 30.2), 17.1 (d, Pb; 2JPP = 30.2) |
CDCl3 |
110 |
| 136D-UM |
21.4 (d, Pa; 2JPP = 30.5), 17.7 (d, Pb; 2JPP = 30.5) |
CDCl3 |
110 |
| 137D-UM |
38.1 (d, Pa; 2JPP = 73.8), 29.3 (d, Pb; 2JPP = 73.8) |
CDCl3 |
110 |
| 138D-UM |
38.1 (d, Pa; 2JPP = 71.1), 29.8 (d, Pb; 2JPP = 71.1) |
CDCl3 |
110 |
| 139D-UM |
37.6 (d, Pa; 2JPP = 77.0), 29.4 (d, Pb; 2JPP = 77.0) |
CDCl3 |
110 |
| 140D-UM |
43.8 (d, Pa; 2JPP = 61.0), 31.8 (d, Pb; 2JPP = 61.0) |
CDCl3 |
110 |
| 141D-UM |
42.8 (d, Pa; 2JPP = 61.0), 31.8 (d, Pb; 2JPP = 61.0) |
CDCl3 |
110 |
| 142D-UM |
44.4 (d, Pa; 2JPP = 63.6), 31.1 (d, Pb; 2JPP = 63.6) |
CDCl3 |
110 |
| 143D-UUM |
28.5 (d, Pa, 2Jab, 32.6), 17.3 (ddd, Pb, 2Jbd, 398.8, 2Jab, 32.6, 2Jbc,14.2), −23.0 (dd, Pc, 2Jcd, 69.2, 2Jbc, 12.2), −29.4 (dd, Pd, 2Jbd, 398.8, 2Jcd, 69.2) |
CDCl3 |
110 |
| 144D-UUM |
39.6 (d, Pa, 2Jab, 86.5), 22.6 (ddd, Pb, 2Jbd, 399.7, 2Jab, 86.5, 2Jbc, 20.3), −8.6 (d, Pd, 2Jbd, 399.7), −10.2 (d, Pc, 2Jbc, 20.3) |
CDCl3 |
110 |
| 145D-UUM |
26.1 (d, Pa, 2Jab, 33.6), 13.1 (ddd, Pb, 2Jbd, 394.2, 2Jab, 33.6, 2Jbc, 22.9), −7.5 (dd, Pd, 2Jbd, 394.2, 2Jcd, 5.1), −8.6 (dd, Pc, 2Jbc, 22.9, 2Jcd, 5.1) |
CDCl3 |
110 |
| 146D-UUM |
24.8 (d, Pa,2Jab, 33.1), 12.5 (ddd, Pb, 2Jbd, 394.2, 2Jab, 33.1, 2Jbc, 10.2), −13.7 (dd, Pc, 2Jcd, 38.1, 2Jbc, 10.2), −16.6 (dd, Pd, 2Jbd, 394.2, 2Jcd, 38.1) |
CDCl3 |
110 |
| 147D-UUM |
45.7 (d, Pa, 2Jab, 68.7), 24.4 (ddd, Pb, 2Jbd, 389.1, 2Jab, 68.7, 2Jbc, 7.6), −29.2 (dd, Pd,2Jbd, 389.1, 2Jcd, 73.8), −33.0 (dd, Pc,2Jcd, 73.6, 2Jbc, 7.6) |
CDCl3 |
110 |
| 148D-UUM |
66.3 (d, Pd, 2Jbd, 369.7), 62.0 (d, Pc, 2Jbc, 22.9), 40.3 (d, Pa, 2Jab, 86.5), 22.8 (ddd, Pb 2Jbd, 396.7, 2Jab, 86.5, 2Jbc, 22.9) |
CDCl3 |
110 |
| 149D-UUM |
61.0 (d, Pd, 2Jbd, 368.8), 58.7 (d, Pc, 2Jbc, 17.8), 42.8 (d, Pa, 2Jab, 61.0), 26.9 (ddd, Pb, 2Jbd, 368.8, 2Jab, 61.0, 2Jbc, 17.8) |
CDCl3 |
110 |
| 150D-UUM |
39.2 (d, Pa, 2Jab, 83.9), 22.7 (ddd, Pb, 2Jbd, 399.3, 2Jab, 83.9, 2Jbc, 20.3), −8.88 (d, Pd, 2Jbd, 399.3), −10.4 (d, Pc, 2Jbc, 20.3) |
CDCl3 |
110 |
| 151D-UUM |
50.8 (d, Pa, 2Jab, 66.1), 30.4 (ddd, Pb, 2Jbd, 394.2, 2Jab, 66.1, 2Jbc, 22.9), −8.2 (d, Pd, 2Jbd, 394.2), −13.1 (d, Pc, 2Jbc, 22.9) |
CDCl3 |
110 |
| 153M |
39.1 |
CDCl3 |
154 |
38.2M |
CDCl3 |
112 |
| 155M |
39.3 |
CDCl3 |
156 |
39.1M |
CDCl3 |
112 |
| 159M |
40.4 |
CDCl3 |
160 |
40.3M |
CDCl3 |
112 |
|
| Platinum(II) |
| 169M |
22.7 (JP–Pt = 3880) |
CDCl3 |
170M |
23.0 (JP–Pt = 3854) |
CDCl3 |
115 |
| 171M |
22.8 (JP–Pt = 3857) |
CDCl3 |
172M |
21.7 (JP–Pt = 4071) |
CDCl3 |
115 |
| 173M |
21.7 (JP–Pt = 4107) |
CDCl3 |
174M |
17.6 (JP–Pt = 3869) |
CDCl3 |
115 |
| 175D |
11.6 (JP–Pt = 3943) |
CDCl3 |
176D |
17.7 (JP–Pt = 3843) |
CDCl3 |
115 |
| 177D |
13.3 (JP–Pt = 3941) |
CDCl3 |
178D |
15.2 (JP–Pt = 3813) |
CDCl3 |
115 |
| 179D |
15.4 (JP–Pt = 3819) |
CDCl3 |
180D |
15.2 (JP–Pt = 3815) |
CDCl3 |
115 |
| 181D |
13.4 (JP–Pt = 3870) |
CDCl3 |
|
|
CDCl3 |
115 |
| 193M |
16.60 (JP–Pt = 3743) |
CDCl3 |
194M |
16.65 (JP–Pt = 3743) |
CDCl3 |
117 |
| 195D |
12.56 (JP–Pt = 3693) |
CDCl3 |
196D |
8.11 (JP–Pt = 3672) |
CDCl3 |
117 |
| 197D |
7.33 (JP–Pt = 3693) |
CDCl3 |
198D |
12.56 (JP–Pt = 3743) |
CDCl3 |
117 |
| 199D |
8.14 (JP–Pt = 3662) |
CDCl3 |
200D |
7.28 (JP–Pt = 3680.4 Hz) |
CDCl3 |
117 |
| 201M-P |
6.15d (2JPP = 71.2, JP–Pt = 3723); −27.9d (2JPP = 71.2) |
CDCl3 |
117 |
| 202M-P |
23.0d (2JPP = 76.3, JP–Pt = 3784); −12.55d (2JPP = 76.3) |
CDCl3 |
117 |
| 203M-P |
3.70d (2JPP = 73.2, JP–Pt = 3717); −30.40d (2JPP = 73.2) |
CDCl3 |
117 |
| 204M-P |
23.1d (2JPP = 76.2, JP–Pt = 3684); −12.5d, 2JPP = 76.2) |
CDCl3 |
117 |
| 205D-UM |
8.75d (2JPP = 25.4, JP–w = 245.7); 2.87 (d, 2JPP = 25.4; JP–Pt = 3784) |
CDCl3 |
117 |
| 206D-UM |
6.27d (2JPP = 24.9, JP–w = 246), 0.40d (2JPP = 24.9, JP–Pt = 3772) |
CDCl3 |
117 |
| 231M-UP |
−115.5, −82.8, −55.0b |
232M-UP |
−133.8, −116.5, −99.5, −72.9b |
CDCl3 |
122 |
13C NMR spectroscopy. The 13C NMR data for diagnostic carbon atoms (C8, C2, C1 and C3) are listed in Table 4. Here C8 carbon is bonded to metal center, C2 is azomethine carbon bonded to coordinating N3 (–C2N3–), C1 carbon is again close to coordinating sulfur atom and finally C3 is ipso carbon of the ring. These carbons undergo shifts to high or low field relative to the uncoordinated ligand and is demonstrated with one example. The C8, C2, and C3 signals of oligomer 1 at 148.0, 156.2 and 132.1 ppm are significantly down field relative to the free ligand at (129.2, 142.0 and 127.2 ppm respectively) except C1 signal at 167.9 ppm which is upfield relative to the free ligand signal at 177.9 ppm. This upfield shift implies greater shielding of C1 carbon when sulfur binds to metal. These trends are general and deviation from the trend can not be ruled out and the reader is advised to look to the original literature for rigorous comparison. The values of the chemical shifts for each category of these carbons fall in a fairly wide range owing to the divergence in the nature of thio-ligands as well as co-ligands forming the complexes listed in Table 4.
31P NMR spectroscopy. The reported 31P NMR data of phosphine containing complexes is placed in Table 5. For more comprehensive understanding of the data given in this Table, compounds are grouped into various categories. Mononuclear complexes containing one monodenate phosphine each (M: 30–32, 35–38, 127,128,153–160,169–174, 193,194) showed one 31P NMR signal. Likewise dinuclear complexes having equivalent P,P-bridging (D: 12, 13, 39–44, 58–82, 103–120, 129–134, 175–181, 195–200) also showed one signal each. Mononuclear complexes with pendant PPh2 group (M-P: 45–48, 83–90, 121, 122, 201–204) have two non-equivalent P nuclei, Pa, Pb and thus showed two doublets involving Pa–Pb coupling. The coordinated Pa gave doublet in the low field region while the pendant Pb showed doublet in the high field region. The mononuclear complexes (M-C: 91–94) with chelating dppm donors but facing different thio-ligand donors showed two doublets in the low field region with small Pa–Pb coupling. The dinuclear complexes (D-UM: 95–98, 135–142, 205, 206) with diphosphines bridging unequal metals, again showed a pair of doublets with pattern similar to M-C category of complexes. The dinuclear complexes with unsymmetrical phosphines bonded to unequal metals (D-UUM: 143–151) have four types of P nuclei and four signals involving various couplings between Pa, Pb, Pc, Pd nuclei were observed. Finally mononuclear complexes with unequal phosphines (M-UP: 231, 232) showed different 3 or 4 31P NMR signals (Chart 4).
 |
| | Chart 4 Bonding modes of phopshines in complexes. M-mononuclear, D -dinuclear, M-P: mononuclear with pendant PPh2 group, M-C: mononuclear with chelating dppm donors facing different donors bonded to metal, D-UM: dinuclear with phosphines bridging unequal metals, D-UUM: dinuclear with unsymmetrical phosphines bonded to unequal metals, M-UP: mononuclear with unequal phosphines. | |
3.5 Factors controlling cylometallation
This survey reveals that thiosemicarbazones (R1R2CN–NH–C(S)–NR3R4) have cyclometallated palladium(II), platinum(II), ruthenium(II), rhodium(III) and iridium(III) only – a group of soft Lewis acids. There is no other metal ion which has formed similar direct M–C (thiosemicarbazone) bond. The R1 groups at C2 carbon undergoing cyclometallation are phenyl rings (with or without a substituent, e.g. MeO, F, etc.), furan, thiopheneyl and pyridyl rings. The R2 group is H, Me or Et as the most common ones and NR3R4 group may be –NH2, NHR3 or NR2 type. Only R1 group has formed M–C bonds and among R2, R3 and R4 groups, only R2 promoted cyclometallation. Among several cyclometallated compounds, there are only a few examples in which cases R1 was phenyl group with no substitution and R2 was hydrogen (Pd:101 19; Pt:116 188; Ru:118,119,122 209, 214, 232; Rh:123 235, 241; Ir:124 246, 251). The cyclometallation by phenyl ring (R1) can be categorized, in relation to thiosemicarbazones, into four different types when: (i) R1 has one or two substituents with R2 being hydrogen, (ii) R1 has no substituent but R2 is methyl, ethyl or phenyl, (iii) R1 has substituents and R2 is methyl, ethyl or phenyl and (iv) R and R2 are hydrogens. Chart 5 shows, in summary, metallation process with change of R and R2 substituents.
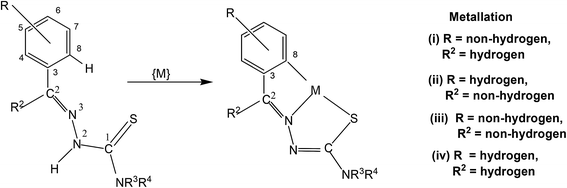 |
| | Chart 5 Metallation with change of R and R2 groups. | |
As regards metallation by furan (R1) and thiopheneyl (R1) rings, there is no report where rings are having substituents, but R2 group must be other than hydrogen for cyclometallation of these heterocyclic rings-based on available information. However, with this category of thio-ligands there are limited examples with PdII (R1 = thiopheneyl group: 159, 160),112 PtII (R1 = furan group, 191–206)117 and RuII (R1 = thiopheneyl, 231, 232)122 and none with RhIII and IrIII. Ruthenium(II) complexes 228–230 have single and double metallation by the pyridyl ring of bis-thiosemicarbazones (L87),121 but there is methyl substitution (R2) at C2 carbon which appears necessary for metallation. It may be noted that there is only one example (ligand L11, complex 14)99 where methyl group present at a position ortho (C4) to ipso (C3) underwent cyclometallation and not the phenyl ring.
It is pertinent to bring forward here some factors supporting cyclometallation. The presence of R substituents in phenyl ring (R1) alter the electron density on aromatic ring via their electromeric or inductive effects which enhance the possibility of activation of C–H bond leading to cyclometallation. This pertains to cases (i) and (iii) as listed in Chart 5. When phenyl ring (R1) has R substituent as hydrogen and R2 is also hydrogen (case iv), there are only a few examples where metallation has occurred (Pd:101 19; Pt:116 188; Ru:118,119,122 209, 214, 232; Rh:123 235, 241; Ir:124 246, 251). Thus when phenyl ring (R1) has R substituent as hydrogen, for causing metallation R2 must be non-hydrogen (case ii) and this has led to formation of several cyclometallated complexes. This is true also in case of thiosemicarbzones where R1 groups are thiopheneyl, furan or pyridyl, requiring R2 as non-hydrogen moiety excepting RuII complex 231 (R2 = H) (vide infra). The effect of methyl substituent at C2 carbon in making thiopheneyl group to metallate is shown in Chart 6 using an example of Pd metal complex. Thiophen-2-formaldehyde thiosemicarbazone (R2 = H) yielded complexes A and B with no metallation,112 but the presence of methyl group at C2 carbon assisted metallation (159–160) by way of enhancing Lewis basicity of N3 nitrogen and also posing steric effect as shown in Chart 6. Chart 7 displays metallation shown by dppm in RuII chemistry forming complexes 231 and 232.122 The smaller bite of dppm assisted metallation by thiopheneyl and phenyl rings in unusual way, as in contrast, dppe merely formed non-metallated complexes.122
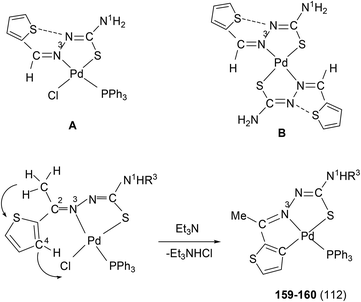 |
| | Chart 6 | |
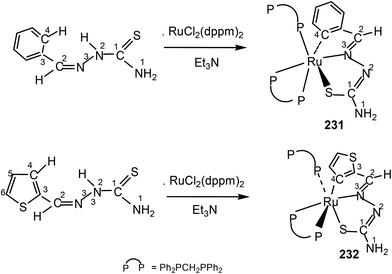 |
| | Chart 7 Metallation caused by dppm in unusual way. | |
4. Applications – biological and catalysis
4.1 Biological applications
Among thio-ligands L39 and L40 and their complexes 123, 124, 127–134, only mononuclear PdII complexes 127 and 128 were most effective antimalarial agents against two Plasmodium falciparum strains, 3D7 (chloroquine sensitive) and K1 (chloroquine and pyrimethamine resistant).109 Tetranuclear complexes 4(Pd) and 182(Pt) showed higher cytotoxic activity in tumor cell lines resistant to cis-platin and etoposide. Other complexes (Pd: 5–8; Pt: 183–185) had a lower cytotoxic values, but are more active than cis-platin and other clinically used drugs.25a,96,97 Complexes 186 to 190 showed cytotoxicity against two human tumor cell lines (viz. HL-60 and U-937), and some are more active than cis-platin.116
4.2 Catalysis
Complex [Pd(C,N,S-L16-2H)(PPh3)] 19 has been found to be excellent catalyst for Suzuki type C–C coupling which involved reactions of aryl halides with phenylboronic acids (eqn (2)).101 The R1 ring substituents of aryl halides used were –C(O)CH3, –CHO and –CN with R2 substituents of phenylboronic acids being H, OCH3 or Cl. The C–X bonds activated were C–Cl, C–Br and C–I. Complexes [Pd(C,N,S-L14-2H)(PPh3)] 17 and [Pd(C,N,S-L18-2H)(PPh3)] 21 showed similar activity as tested for C–I activation with R1 = –C(O)CH3, R2 = H.
Complex 19 has also been investigated for catalytic activity towards Buchwald type C–N coupling reactions between aryl halides (X = I, Br tested) (eqn (3)) and primary/secondary amines (Chart 8). It was found that this complex exhibited ten times higher TON for X = I over X = Br. This catalyst showed slightly better catalytic efficiency than bis-complex [Pd(N3,S-L16-H)2].106 The TON was low for C–Cl bond activation as tested for bis-complex for all the amines used (Chart 8). Both types of reactions investigated have either better or comparable catalytic activity to that reported in literature. A special feature is efficient activation of C–Cl bonds which is rather scarce.125–129
| |
 | (2) |
 |
| | Chart 8 Amines/secondary amines. | |
Complexes 152 and 157 have been tested as catalyst for the Suzuki coupling of phenylboronic acid with p-haloacetophenones.113 Here activation of C–I, C–Br, C–Cl, and C–F bonds was performed (X = I, Br, Cl, F, eqn (4)). It may be added here that complex, [PdCl(N3,S-btsc)(PPh3)] 158 (btsc is anion of benzaldehyde thiosemicarbazone chelating to Pd center via its N,S-donor atoms) also showed similar activity but efficiency was somewhat low relative to organometallic complexes 152 and 157, especially in terms of time taken to complete the reaction.113 The activation of C–Cl bonds are industrially significant due to easy availability of the relatively inexpensive arylchlorides and also the activation of C–F bonds though less efficient is scarce.113 In addition to C–C coupling, C–N coupling is also observed using complexes 152, 157 and [PdCl(N3,S-btsc)(PPh3)] 158 (eqn (5) and (6)). However there was no activation of C–F bond. The trend in C–N bond formation was similar to that with primary amines, however turn over numbers were lower in case of secondary amines (eqn (6)) relative to the primary amines (eqn (5)).113
| |
 | (3) |
| |
 | (4) |
| |
 | (5) |
| |
 | (6) |
5. Conclusion
An interesting feature of reactions of thiosemicarbazones with PdII/PtII, in the absence of co-ligands such as tertiary phosphines, has yielded self-assembled cyclometallated tetranuclear oligomers. These oligomers on further reactions with tertiary phosphines yielded mono- and di-nuclear complexes which have seven different types of bonding modes displayed by these phosphines (Chart 4). The M–C bonds formed by thiosemicarbazones appear to be inert and remain intact even when phosphines react with oligomers. However the facile aerial oxidation of the thiolate-sulfur of the coordinated thiosemicarbazone to sulfinate formed rhodium complexes (233 to 237). Here it was shown by theoretical studies this oxidation is attributed to the presence of the hydride ligand in the complexes, which makes the sulfur atom electron rich and susceptible to oxidation by molecular oxygen. The presence of electronegative chloride instead of hydride (239–243) reduces electron density on the sulfur atom making its spontaneous aerial oxidation impossible.130 A few complexes (Pd/Pt) tested for anti-malarial and anti-cancer activity have shown a big scope for developing this area for antimicrobial, anti-malarial and anti-cancer activity. Further, a few Pd complexes tested for catalysis have emerged excellent catalyst for Suzuki type C–C coupling which involved reactions of aryl halides/p-haloacetophenones with phenylboronic acids via activation of C–F, C–Cl, C–Cl and C–I bonds. Also a few complexes have been investigated for catalytic activity towards Buchwald type C–N coupling reactions between aryl halides and primary/secondary amines. The activation of C–Cl bonds are industrially significant due to easy availability of the relatively inexpensive arylchlorides and also the activation of C–F bonds though less efficient is scarce. Thus catalysis provides a useful area of investigations. This review provides a summary of coordination modes of phosphines in complexes and is rich in NMR spectroscopy, especially 31P NMR. The cyclometallation area encompassing thiosemicarbazones provides a promising area of research specially with respect to biological applications and catalysis.
Acknowledgements
This author thanks RSC advances for invitation to write review on metallation by thiosemi-carbazones and the Council of Scientific and Industrial Research (CSIR), New Delhi for Emeritus Scientist position vide Scheme no. [21(0904)/12-EMR-II].
References
- I. Omae, Organometallic Intramolecular-coordination Compounds, Elsevier Science, Amsterdam, 1986 Search PubMed.
- G. R. Newkome, W. E. Puckett, W. K. Gupta and G. E. Kiefer, Chem. Rev., 1986, 86, 451–489 CrossRef CAS.
- A. D. Ryabov, Chem. Rev., 1990, 90, 403–424 CrossRef CAS.
- P. Espinet, M. A. Esteruelas, L. A. Oro, J. L. Serrano and E. Sola, Coord. Chem. Rev., 1992, 117, 215–274 CrossRef CAS.
- B. S. Wild, Coord. Chem. Rev., 1997, 166, 291–311 CrossRef.
- L. Main and B. K. Nicholson, Adv. Met.-Org. Chem., 1994, 3, 1–50 CAS.
- J. Spencer and M. Pfeffer, Adv. Met.-Org. Chem., 1998, 6, 103–144 CAS.
- J. Dopont, M. Pfeffer and J. Spencer, Eur. J. Inorg. Chem., 2001, 1917–1927 CrossRef.
- L. Botella and C. Najera, Angew. Chem., Int. Ed., 2002, 41, 179–181 CrossRef CAS.
- J. Albert, J. M. Cadena, J. R. Granell, X. Solans and M. Font-Bardia, Tetrahedron: Asymmetry, 2000, 11, 1943–1955 CrossRef CAS.
- D. Song, Q. Wu, A. Hook, I. Kozin and S. Wang, Organometallics, 2000, 20, 4683–4689 CrossRef.
- V. A. Kozlov, D. V. Aleksanyan, Y. V. Nelyubina, K. A. Lyssenko, E. I. Gutsul, L. N. Puntus, A. A. Vasil'ev, P. V. Petrovskii and I. L. Odinets, Organometallics, 2008, 27, 4062–4070 CrossRef CAS.
- M. Curic, D. Babic, A. Visnjevac and K. Molcanov, Inorg. Chem., 2005, 44, 5975–5977 CrossRef CAS PubMed.
- P. Steenwinkel, R. A. Gossage and G. Van Koten, Chem.–Eur. J., 1998, 4, 759–762 CrossRef CAS.
- M. Albrecht and G. van Kotten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS.
- M. Pfeffer, J. P. Sutter, M. A. Rottevel, A. de Cian and J. Fischer, Tetrahedron, 1992, 48, 2427–2440 CrossRef CAS.
- A. D. Ryabov, R. van Eldik, G. Le Borgne and M. Pfeffer, Organometallics, 1993, 12, 1386–1393 CrossRef CAS.
- A. Fernández, D. Vázquez-García, J. J. Fernández, M. López-Torres, A. Suárez, A. Castro-Juiz, J. M. Ortigueira and J. M. Vila, New J. Chem., 2002, 26, 105–112 RSC.
- A. Bose and C. H. Saha, J. Mol. Catal., 1989, 49, 271–283 CrossRef CAS.
- S. Y. M. Chooi, P. H. Leung, C. C. Lim, K. F. Mok, G. H. Quek, K. Y. Sim and M. K. Tan, Tetrahedron: Asymmetry, 1992, 3, 529–532 CrossRef CAS.
- C. Navarro-Ranninger, I. López-Solera, V. M. Gonzalez, J. M. Pérez, A. Alvarez-Valdés, A. Martin, P. R. Raithby, J. R. Masaguer and C. Alonso, Inorg. Chem., 1996, 35, 5181–5187 CrossRef CAS.
- W. A. Herrmann, C. Brossmer, K. Oefele, C. P. Reisinger, T. Priermeier, M. Beller and H. Fischer, Angew. Chem., Int. Ed. Engl., 1995, 34, 1844–1848 CrossRef CAS PubMed.
- M. López-Torres, A. Fernández, J. J. Fernández, S. Castro-Juiz, A. Suárez, J. M. Vilay and M. T. Pereira, Organometallics, 2001, 20, 1350–1353 CrossRef.
- C. Navarro-Ranninger, I. López Solera, J. Rodríguez, J. L. García-Ruano, P. R. Raithby, J. R. Masaguer and C. Alonso, J. Med. Chem., 1993, 36, 3795–3801 CrossRef CAS.
-
(a) A. G. Quiroga and C. Navarro-Ranninger, Coord. Chem. Rev., 2004, 248, 119–133 CrossRef CAS PubMed;
(b) A. Garoufis, S. K. Hadjikakou and N. Hadjiliadis, Coord. Chem. Rev., 2009, 253, 1384–1397 CrossRef CAS PubMed;
(c) J. L. Hickey and P. S. Donnelly, Coord. Chem. Rev., 2012, 256, 2367–2380 CrossRef CAS PubMed.
- T. S. Lobana, R. Sharma, G. Bawa and S. Khanna, Coord. Chem. Rev., 2009, 253, 977–1055 CrossRef CAS PubMed.
- T. S. Lobana, Rekha and R. J. Butcher, Transition Met. Chem., 2004, 29, 291–295 CrossRef CAS.
- T. S. Lobana, Rekha, R. J. Butcher, A. Castineiras, E. Bermejo and P. V. Bharatam, Inorg. Chem., 2006, 45, 1535–1542 CrossRef CAS PubMed.
- E. M. Jouad, A. Riou, M. Allain, M. A. Khan and G. M. Bouet, Polyhedron, 2001, 20, 67–74 CrossRef CAS.
- S. Lhuachan, S. Siripaisarnpipat and N. Chaichat, Eur. J. Inorg. Chem., 2003, 263–267 CrossRef CAS PubMed.
- E. Bermejo, R. Carballo, A. Castineiras, R. Dominguez, C. Marchle-Moessmer, J. Strahle and D. X. West, Polyhedron, 1999, 18, 3695–3704 CrossRef CAS.
- P. Gomez-Saiz, J. Garcia-Tojal, M. A. Maestro, J. Mahla, F. J. Arniaz, L. Lezama and T. Rojo, Eur. J. Inorg. Chem., 2003, 2639–2650 CrossRef CAS PubMed.
- E. Labisbal, K. D. Haslow, A. Sousa-Pedrares, J. Valdes-Martinez, S. Harnandez-Ortega and D. X. West, Polyhedron, 2003, 22, 2831–2837 CrossRef CAS.
- D. Kovala-Demertzi, M. A. Demertzis, J. Valdez-Martinez, S. Hernandez-Ortega, G. Espinosa-Perez, D. X. West, M. M. Salberg, G. A. Bain and P. D. Bloom, Polyhedron, 1996, 15, 2587–2596 CrossRef CAS.
- J. S. Casas, E. E. Castellano, M. C. Rodriguez-Arguelles, A. Sanchez, J. Sordo and J. Zukerman-Schpector, Inorg. Chim. Acta, 1997, 280, 183–188 CrossRef.
- J. Garcia-Tojal, J. L. Pizarro, A. Garcia-Orad, A. R. Perez-Sanz, M. Ugalde, A. A. Diaz, J. L. Serra, M. I. Arriortua and T. Rojo, J. Inorg. Biochem., 2001, 86, 627–633 CrossRef CAS.
- J. S. Casas, M. V. Castano, M. C. Cituenyes, A. Sanchez and J. Sordo, Polyhedron, 2002, 21, 1651–1660 CrossRef CAS.
- T. S. Lobana, G. Bawa, A. Castineiras and R. J. Butcher, Inorg. Chem. Commun., 2007, 10, 506–509 CrossRef CAS PubMed.
- D. Kovala-Demertzi, N. Kourkoumelis, M. A. Demertzis, J. R. Miller, C. S. Frampton, J. K. Swearingen and D. X. West, Eur. J. Inorg. Chem., 2000, 727–734 CrossRef CAS.
- Z. Lu, C. White, A. L. Rheingold and R. H. Crabtree, Inorg. Chem., 1993, 32, 3991–3994 CrossRef CAS.
- T. S. Lobana, G. Bawa, R. J. Butcher, B.-J. Liaw and C. W. Liu, Polyhedron, 2006, 25, 2897–2903 CrossRef CAS PubMed.
- L. J. Ashfield, A. R. Cowley, J. R. Dilworth and P. S. Donnelly, Inorg. Chem., 2004, 43, 4121–4123 CrossRef CAS PubMed.
- I. Pal, F. Basuli, T. C. W. Mak and S. Bhattacharya, Angew. Chem., Int. Ed., 2001, 40, 2923–2925 CrossRef CAS.
- R. B. de Oliveira, E. M. de Souza-Fagundes, R. P. P. Soares, A. A. Andrade, A. U. Krettli and C. L. Zani, Eur. J. Med. Chem., 2008, 43, 1983–1988 CrossRef CAS PubMed.
- K. J. Duffy, A. N. Shaw, E. Delorme, S. B. Dillon, C. Erickson-Miller, L. Giampa, Y. Huang, R. M. Keenan, P. Lamb, N. Liu, S. G. Miller, A. T. Price, J. Rosen, H. Smith, K. J. Wiggall, L. Zhang and J. I. Luengo, J. Med. Chem., 2002, 45, 3573–3575 CrossRef CAS PubMed.
- M. Abid, S. M. Agarwal and A. Azam, Eur. J. Med. Chem., 2008, 43, 2035–2039 CrossRef CAS PubMed.
- A. Walcourt, M. Loyevsky, D. B. Lovejoy, V. R. Gordeuk and D. R. Richardson, Int. J. Biochem. Cell Biol., 2004, 36, 401–407 CrossRef CAS.
- N. Fujii, J. P. Mallari, E. J. Hansell, Z. Mackey, P. Doyle, Y. M. Zhou, J. Gut, P. J. Rosenthal, J. H. McKerrow and R. K. Guy, Bioorg. Med. Chem. Lett., 2005, 15, 121–123 CrossRef CAS PubMed.
- X. Du, C. Guo, E. Hansel, P. S. Doyle, C. R. Caffrey, T. P. Holler, J. H. McKerrow and F. E. Cohen, J. Med. Chem., 2002, 45, 2695–2707 CrossRef CAS PubMed.
- S. A. Khan and M. Yusuf, Eur. J. Med. Chem., 2009, 44, 2597–2600 CrossRef CAS PubMed.
- S. A. Khan, P. Kumar, R. Joshi, P. F. Iqbal and K. Saleem, Eur. J. Med. Chem., 2008, 43, 2029–2034 CrossRef CAS PubMed.
- C. Costello, T. Karpanen, P. A. Lambert, P. Mistry, K. J. Parker, D. L. Rathbone, J. Ren, L. Wheeldon and T. Worthington, Bioorg. Med. Chem. Lett., 2008, 18, 1708–1711 CrossRef CAS PubMed.
- D. Kovala-Demertzi, M. A. Demertzis, E. Filou, A. A. Pantazaki, P. N. Yadav, J. R. Miller, Y. Zheng and D. A. Kyriakidis, Biometals, 2003, 16, 411–418 CrossRef CAS.
- J. M. Kolesar, W. R. Schelman, P. G. Geiger, K. D. Holen, A. M. Traynor, D. B. Alberti, J. P. Thomas, C. R. Chitambar, G. Wilding and W. E. Antholine, J. Inorg. Biochem., 2008, 102, 693–698 CrossRef CAS PubMed.
- M.-C. Liu, T.-S. Lin and A. C. Sartorelli, J. Med. Chem., 1992, 35, 3672–3677 CrossRef CAS.
- I. Dilovic, M. Rubcic, V. Vrdoljak, S. K. Pavelic, M. Kralj, I. Piantanida and M. Cindric, Bioorg. Med. Chem., 2008, 16, 5189–5198 CrossRef CAS PubMed.
- A. Maiti, A. K. Guha and S. Ghosh, J. Inorg. Biochem., 1988, 33, 57–65 CrossRef CAS.
- D. Singh and R. V. Singh, J. Inorg. Biochem., 1993, 50, 227–234 CrossRef CAS.
- N. C. Kasuga, K. Sekino, C. Koumo, N. Shimada, M. Ishikawa and K. Nomiya, J. Inorg. Biochem., 2000, 84, 55–65 CrossRef.
- C. R. Kowol, R. Eichinger, M. A. Jakupec, M. Galanski, V. B. Arion and B. K. Keppler, J. Inorg. Biochem., 2007, 101, 1946–1957 CrossRef CAS PubMed.
- M. B. Ferrari, F. Bisceglie, G. Pelosi, M. Sassi, P. Tarasconi, M. Cornia, S. Capacchi, R. Albertini and S. Pinelli, J. Inorg. Biochem., 2002, 90, 113–126 CrossRef.
- W. E. Antholine, J. M. Knight and D. H. Petering, J. Med. Chem., 1976, 19, 339–341 CrossRef CAS.
- Z. Afrasiabi, E. Sinn, W. Lin, Y. Ma, C. Campana and S. Padhye, J. Inorg. Biochem., 2005, 99, 1526–1531 CrossRef CAS PubMed.
- J. S. Casas, M. V. Castano, M. C. Cifuentes, J. C. Garcia-Monteagudo, A. Sanchez, J. Sordo and U. Abram, J. Inorg. Biochem., 2004, 98, 1009–1016 CrossRef CAS PubMed.
- B. Garcia, J. Garcia-Tojal, R. Ruiz, R. Gil-Garcia, S. Ibeas, B. Donnadieu and J. M. Leal, J. Inorg. Biochem., 2008, 102, 1892–1900 CrossRef CAS PubMed.
- M. B. Ferrari, S. Capacchi, G. Pelosi, G. Reffo, P. Tarasconi, R. Albertini, S. Pinelli and P. Lunghi, Inorg. Chim. Acta, 1999, 286, 134–141 CrossRef.
- J. P. Scovill, D. L. Klayman and C. F. Franchino, J. Med. Chem., 1982, 25, 1261–1264 CrossRef CAS.
- J. Patole, S. Padhye, M. S. Moodbidri and N. Shirsat, Eur. J. Med. Chem., 2005, 40, 1052–1055 CrossRef CAS PubMed.
- D. Kovala-Demertzi, A. Boccarelli, M. A. Demertzis and M. Coluccia, Chemotherapy, 2004, 53, 148–152 CrossRef PubMed.
- P. Chellan, N. Shunmoogam-Gounden, D. T. Hendricks, J. Gut, P. J. Rosenthal, C. Lategan, P. J. Smith, K. Chibale and G. S. Smith, Eur. J. Inorg. Chem., 2010, 3520–3528 CrossRef CAS PubMed.
- D. Kovala-Demertzi, M. A. Demertzis, J. R. Miller, C. Papadopoulou, C. Dodorou and G. Filousis, J. Inorg. Biochem., 2001, 86, 555–563 CrossRef CAS.
- Z. Lakovidou, A. Papageorgiou, M. A. Demertzis, E. Mioglou, D. Mourelatos, A. Kotsis, P. N. Yadav and D. Kovala-Demertzi, Anticancer Drugs, 2001, 12, 65–70 CrossRef CAS PubMed.
- D. Kovala-Demertzi, P. N. Yadav, M. A. Demertzis and M. Coluccia, J. Inorg. Biochem., 2000, 78, 347–354 CrossRef CAS.
- A. M. Asiri and S. A. Khan, Molecules, 2010, 15, 4784–4791 CrossRef CAS PubMed.
- T. Rosu, E. Pahontu, S. Pasculescu, R. Georgescu, N. Stanica, A. Curaj, A. Popescu and M. Leabu, Eur. J. Med. Chem., 2010, 45, 1627–1634 CrossRef CAS PubMed.
- S. A. Khan and M. Yusuf, Eur. J. Med. Chem., 2009, 44, 2270–2274 CrossRef CAS PubMed.
- M. Vieites, L. Otero, D. Santos, C. Olea-Azar, E. Norambuena, G. Aguirre, H. Cerecetto, M. González, U. Kemmerling, A. Morello, J. D. Maya and D. Gambino, J. Inorg. Biochem., 2009, 103, 411–418 CrossRef CAS PubMed.
- L. Otero, C. Folch, G. Barriga, C. Rigol, L. Opazo, M. Vieites, D. Gambino, H. Cerecetto, E. Norambuena and C. Olea-Azar, Spectrochim. Acta, Part A, 2008, 70, 519–523 CrossRef PubMed.
- S. Padhye, Z. Afrasiabi, E. Sinn, J. Fok, K. Mehta and N. Rath, Inorg. Chem., 2005, 44, 1154–1156 CrossRef CAS PubMed.
- K. S. Ferraz, L. Ferandes, D. Carrilho, M. C. Pinto, F. Leite Mde, E. M. Souza-Fagundes, N. L. Speziali, I. C. Mendes and H. Beraldo, Bioorg. Med. Chem., 2009, 17, 7138–7144 CrossRef CAS PubMed.
- A. C. Caires, Adv. Anticancer Agents Med. Chem., 2007, 7, 484–491 CrossRef CAS.
- J. S. Casas, E. E. Castellano, J. Ellena, M. S. García-Tasende, M. L. Pérez-Parallé, A. Sánchez, A. Sánchez-González, J. Sordo and A. Touceda, J. Inorg. Biochem., 2008, 102, 33–45 CrossRef CAS PubMed.
- A. I. Matesanz and P. Souza, J. Inorg. Biochem., 2007, 101, 1354–1361 CrossRef CAS PubMed.
- T. S. Lobana, S. Indoria, A. K. Jassal, H. Kaur, D. S. Arora and J. P. Jasinski, Eur. J. Med. Chem., 2014, 76, 145–154 CrossRef CAS PubMed.
- R. K. Mahajan, T. P. S. Walia, Sumanjit and T. S. Lobana, Anal. Sci., 2006, 22, 389–392 CrossRef CAS.
- R. K. Mahajan, I. Kaur and T. S. Lobana, Talanta, 2003, 59, 101–105 CrossRef CAS.
- R. K. Mahajan, T. P. S. Walia, Sumanjit and T. S. Lobana, Talanta, 2005, 67, 755–759 CrossRef CAS PubMed.
- R. K. Mahajan, R. Kaur and T. S. Lobana, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2006, 45, 639–642 Search PubMed.
- D. Nandni, T. S. Lobana and R. K. Mahajan, Anal. Lett., 2009, 42, 2474–2484 CrossRef CAS.
- J. S. Casas, M. S. Garcia-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS.
- J. S. Casas, M. S. Garcia-Tasende and J. Sordo, Coord. Chem. Rev., 1999, 193–195, 283–359 CrossRef CAS.
- D. X. West, S. Padhye and P. B. Sonawane, Struct. Bonding, 1991, 76, 4–49 CrossRef.
- D. X. West, A. E. Liberta, S. B. Padhye, R. C. Chikate, P. B. Sonawane, A. S. Kumbhar and R. G. Yerande, Coord. Chem. Rev., 1993, 123, 49–71 CrossRef CAS.
- S. Padhye and G. B. Kauffman, Coord. Chem. Rev., 1985, 63, 127–160 CrossRef CAS.
- J. M. Vila, T. Pereira, A. Amoedo, M. Grana, J. Martinez, M. Lopez-Torres and A. Fernandez, J. Organomet. Chem., 2001, 623, 176–184 CrossRef CAS.
- A. G. Quiroga, J.
M. Pérez, I. López-Solera, J. R. Masaguer, A. Luque, P. Román, A. Edwards, C. Alonso and C. Navarro-Ranninger, J. Med. Chem., 1998, 41, 1399–1408 CrossRef CAS PubMed.
- A. G. Quiroga, J. M. Pérez, E. I. Montero, C. Alonso and C. Navarro-Ranninger, J. Inorg. Biochem., 1999, 75, 293–301 CrossRef CAS.
- A. Amoedo, L. A. Adrio, J. M. Antelo, J. Martínez, M. Teresa Pereira, A. Fernández and J. M. Vila, Eur. J. Inorg. Chem., 2006, 3016–3021 CrossRef CAS PubMed.
- L. Adrio, J. M. Antelo, J. J. Fernández, K. K. Mimi Hii, M. T. Pereira and J. M. Vila, J. Organomet. Chem., 2009, 694, 747–752 CrossRef CAS PubMed.
- A. Amoedo, M. Grana, J. Martinez, T. Pereira, M. Lopez-Torres, A. Fernandez, J. J. Fernandez and J. M. Vila, Eur. J. Inorg. Chem., 2002, 613–620 CrossRef CAS.
- J. Dutta and S. Bhattacharya, RSC Adv., 2013, 3, 10707–10721 RSC.
- J. M. Vila, M. T. Pereira, J. M. Ortigueira, M. Grana, D. Lata, A. Suarez, J. J. Fernandez, A. Fernandez, M. Lopez-Torres and H. Adams, J. Chem. Soc., Dalton Trans., 1999, 4193–4201 RSC.
- J. Martinez, L. A. Adrio, J. M. Antelo, M. T. Pereira, J. J. Fernandez and J. M. Vila, Polyhedron, 2006, 25, 2848–2858 CrossRef CAS PubMed.
- J. Martinez, M. T. Pereira, I. Buceta, G. Alberdi, A. Amoedo, J. J. Fernandez, M. Lopez-Torres and J. M. Vila, Organometallics, 2003, 22, 5581–5584 CrossRef CAS.
- J. Martınez, L. A. Adrio, J. M. Antelo, J. M. Ortigueira, M. T. Pereira, M. L. Torres and J. M. Vila, J. Organomet. Chem., 2006, 691, 2891–2901 CrossRef PubMed.
- J. Martinez, M. T. Pereira, J. M. Ortigueira, B. Bermudez, J. M. Antelo, A. Fernandez and J. M. Vila, Polyhedron, 2012, 31, 217–226 CrossRef CAS PubMed.
- L. A. Adrio, A. Amoedo, J. M. Antelo, J. J. Fernandez, J. Martınez, J. M. Ortigueira, M. T. Pereira and J. M. Vila, Z. Anorg. Allg. Chem., 2005, 631, 2204–2209 CrossRef CAS PubMed.
- J. Martínez, L. A. Adrio, J. M. Antelo, J. M. Ortigueira, M. T. Pereira, J. J. Fernandez, A. Fernandez and J. M. Vila, J. Organomet. Chem., 2006, 691, 2721–2733 CrossRef PubMed.
- P. Chellan, S. Nasser, L. Vivas, K. Chibale and G. S. Smith, J. Organomet. Chem., 2010, 695, 2225–2232 CrossRef CAS PubMed.
- J. M. Antelo, L. A. Adrio, B. Bermudez, J. Martinez, M. T. Pereira, J. M. Ortigueira, M. L. Torres and J. M. Vila, J. Organomet. Chem., 2013, 740, 83–91 CrossRef CAS PubMed.
- T. S. Lobana, G. Bawa, G. Hundal and M. Zeller, Z. Anorg. Allg. Chem., 2008, 634, 931–937 CrossRef CAS PubMed.
- T. S. Lobana, P. Kumari, R. J. Butcher, T. Akitsu, Y. Aritake, J. Perles, F. J. Fernandez and M. C. Vega, J. Organomet. Chem., 2012, 701, 17–26 CrossRef CAS PubMed.
- P. Paul, P. Sengupta and S. Bhattacharya, J. Organomet. Chem., 2013, 724, 281–288 CrossRef CAS PubMed.
- T. S. Lobana, G. Bawa, A. Castineiras and R. J. Butcher, Inorg. Chem. Commun., 2007, 10, 506–509 CrossRef CAS PubMed.
- D. V. Garcia, A. Fernandez, J. J. Fernandez, M. L. Torres, A. Suarez, J. M. Ortigueira, J. M. Vila and H. Adams, J. Organomet. Chem., 2000, 595, 199–207 CrossRef.
- S. Halder, P. Paul, S. M. Peng, G. H. Lee, A. Mukherjee, S. Dutta, U. Sanyal and S. Bhattacharya, Polyhedron, 2012, 45, 177–184 CrossRef CAS PubMed.
- L. Adrio, J. M. Antelo, J. M. Ortigueira, D. Lata, M. T. Pereira, M. L. Torres and J. M. Vila, Z. Anorg. Allg. Chem., 2007, 633, 1875–1882 CrossRef CAS PubMed.
- N. S. Chowdhury, D. K. Seth, M. G. B. Drew and S. Bhattacharya, Inorg. Chim. Acta, 2011, 372, 183–190 CrossRef CAS PubMed.
- S. Dutta, F. Basuli, A. Castineiras, S. M. Peng, G. H. Lee and S. Bhattacharya, Eur. J. Inorg. Chem., 2008, 4538–4546 CrossRef CAS PubMed.
- R. N. Prabhu, D. Pandiarajan and R. Ramesh, J. Organomet. Chem., 2009, 694, 4170–4177 CrossRef CAS PubMed.
- B. K. Dey, P. Paul and S. Bhattacharya, J. Indian Chem. Soc., 2014, 91, 359–366 CAS.
- T. S. Lobana, G. Bawa, A. Castineiras, R. J. Butcher and M. Zeller, Organometallics, 2008, 27, 175–180 CrossRef CAS.
- R. Acharyya, S. Dutta, F. Basuli, S.-M. Peng, G.-H. Lee, L. R. Falvello and S. Bhattacharya, Inorg. Chem., 2006, 45, 1252–1259 CrossRef CAS PubMed.
- S. Basu, R. Acharya, F. Basuli, S.-M. Peng, G.-H. Lee, G. Mostafa and S. Bhattacharya, Inorg. Chim. Acta, 2010, 363, 2848–2856 CrossRef CAS PubMed.
- P. Paul, S. Datta, S. Halder, R. Acharyya, F. Basuli, R. J. Butcher, S. M. Peng, G. H. Lee, A. Castineiras, M. G. B. Drew and S. Bhattacharya, J. Mol. Catal. A: Chem., 2011, 344, 62–73 CrossRef CAS PubMed.
-
(a) F. Godoy, C. Segarra, M. Poyatos and E. Peris, Organometallics, 2011, 30, 684–688 CrossRef CAS;
(b) R. K. Das, B. Saha, S. M. W. Rahaman and J. K. Bera, Chem.–Eur. J., 2010, 16, 14459–14468 CrossRef CAS PubMed;
(c) H. Firouzabadi, N. Iranpoor and M. Gholinejad, J. Mol. Catal. A: Chem., 2010, 321, 110–116 CrossRef CAS PubMed;
(d) C. F. Fu, C. C. Lee, Y. H. Liu, S. M. Peng, S. Warsink, C. J. Elsevier, J. T. Chen and S. T. Liu, Inorg. Chem., 2010, 49, 3011–3018 CrossRef CAS PubMed;
(e) M. C. Lipke, R. A. Woloszynek, L. Ma and J. D. Protasiewicz, Organometallics, 2009, 28, 188–196 CrossRef CAS.
-
(a) R. Pereira and J. Cvengro, J. Organomet. Chem., 2013, 729, 81–85 CrossRef CAS PubMed;
(b) E. K. Bullough, M. A. Little and C. E. Willans, Organometallics, 2013, 32, 570–577 CrossRef CAS;
(c) M. M. Tamizh, B. F. T. Cooper, C. L. B. Macdonald and R. Karvembu, Inorg. Chim. Acta, 2013, 394, 391–400 CrossRef PubMed;
(d) S. J. Sabounchei, M. Panahimehr, M. Ahmadi, Z. Nasri and H. R. Khavasi, J. Organomet. Chem., 2013, 723, 207–213 CrossRef CAS PubMed;
(e) G. K. Rao, A. Kumar, B. Kumar, D. Kumar and A. Kumar Singh, Dalton Trans., 2012, 41, 1931–1937 RSC;
(f) P. Mao, L. Yang, Y. Xiao, J. Yuan, X. Liu and M. Song, J. Organomet. Chem., 2012, 705, 39–43 CrossRef CAS PubMed;
(g) D. Pandiarajan and R. Ramesh, J. Organomet. Chem., 2012, 708–709, 18–24 CrossRef CAS PubMed;
(h) V. A. Kozlov, D. V. Aleksanyan, Y. V. Nelyubina, K. A. Lyssenko, P. V. Petrovskii, A. A. Vasilev and I. L. Odinets, Organometallics, 2011, 30, 2920–2932 CrossRef CAS.
-
(a) V. Polshettiwar, C. Len and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599–2626 CrossRef CAS PubMed;
(b) A. Fihri, P. Meunier and J. C. Hierso, Coord. Chem. Rev., 2007, 251, 2017–2055 CrossRef CAS PubMed;
(c) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS PubMed.
-
(a) G. Zhang, X. Wang, C. Li and D. Yin, Synth. Commun., 2013, 43, 456–463 CrossRef CAS;
(b) X. D. Fei, Z. Zhou, W. Li, Y. M. Zhu and J. K. Shen, Eur. J. Org. Chem., 2012, 3001–3008 CrossRef CAS PubMed;
(c) X. Hao, J. Yuan, G. A. Yu, M. Q. Qiu, N. F. She, Y. Sun, C. Zhao, S. L. Mao, J. Yin and S. H. Liu, J. Organomet. Chem., 2012, 706–707, 99–105 CrossRef CAS PubMed;
(d) S. Meiries, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 3402–3409 CrossRef CAS.
- D. K. Seth and S. Bhattacharya, J. Organomet. Chem., 2011, 696, 3779–3784 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.