DOI:
10.1039/C5RA04680G
(Paper)
RSC Adv., 2015,
5, 41112-41119
Mechanistic study of the hydrolytic degradation and protonation of temozolomide†
Received
17th March 2015
, Accepted 17th April 2015
First published on 21st April 2015
Abstract
Temozolomide, an anticancer and chemotherapy prodrug, undergoes pH dependent ring-opening under both acidic and alkaline conditions. While the rate of degradation accelerates with an increase in pH, it never comes to a complete halt under acidic conditions. Herein, the ring-opening of temozolomide is investigated, in both neutral and acidic conditions, to find out the energy differences and the effects of an acidic environment on its activation energy. Two possible and different pathways have been considered for the ring-opening reaction. When compared to path-2, the rate-determining step (first TS) for path-1 is about 15 kcal mol−1 more favourable. However, the second path led to a more stable product. It seems that the energy-favoured overall mechanism is a combination of the two paths (tautomerization may occur during the process). Moreover, in order to elucidate the role of the acidic conditions on the increased stability and mechanism of the drug, the protonation of all possible sites was examined. This predicts that the protonation of the oxygen of the amide group is the preferable site for protonation that would stabilize the system by about 1.23 kcal mol−1 more than the next favourable protonation site (protonated nitrogen in the imidazole ring). The low energy barrier (6 kcal mol−1) for proton exchange in these two sites results in the simultaneous existence of both N-protonated and O-protonated structures. In addition, the pKa values were calculated and averaged out at −2.625. This value indicates the extremely acidic feature of the protonated temozolomide that does not protonate or deprotonate over the normal pH range. The protonation and degradation mechanism were treated using density functional theory (B3LYP) and by employing the complete basis set (CBS-4M) method. Moreover, the high-level G3MP2 level was used on some occasions.
Introduction
As an antitumor prodrug, temozolomide (TMZ) has attracted a lot of attention due to its decomposition mechanism and advantages over other alkylating agents (e.g. dacarbazine and mitozolomide). As a prototype of TMZ, mitozolomide (MTZ) exhibits toxic side effects and failed to pass phase II clinical trials.1 Now TMZ is the only alkylating agent of the imidazotetrazine series that is being used for the treatment of a wide spectrum of malignant, gliomas and some types of skin cancers.2–5
The activation mechanism of TMZ has been rationalized by means of NMR spectroscopy.6,7 TMZ tetrazinone ring-opening, which eventually leads to the complete degradation of the system, begins with the addition of a water molecule to the carbonyl moiety. Subsequently, by CO2 elimination, the intermediate bioactive molecule (MTIC) is generated. Thereafter, the MTIC undergoes decomposition and releases the methylating species (methyldiazonium ion) and by-product AIC. The mechanism of degradation is highly pH sensitive. The rate of TMZ degradation increases with an increase in pH.6 The half-life of TMZ in phosphate buffer at physiological pH (7.4) is around 1.83 hours,6 whereas the stability of drug in acidified human plasma (pH < 4) increases to about 24 hour.8
The chemical stability of TMZ has been an issue in prolonged storage. The colors of the pure TMZ crystals fade from white to pink and then light brown during spontaneous degradation. A complete color change in the white crystals of TMZ has been seen after 4 hours for drugs stored at 45% relative humidity.9 In order to prevent the spontaneous degradation and enhance the stability of the drug, several co-crystals of TMZ have been studied.10–13 In addition, nine TMZ polymorphs and a system that extends the drug stability for prolonged storage were reported in the patent literature.9,14 The TMZ co-crystal with anthranilic acid with pKa 4.95 is stable for two weeks, which is one week more when compared to TMZ under the same conditions.10 The stability of the TMZ–saccharin co-crystal (pKa 2.3) is around two months under ambient conditions.11 Moreover, acidified temozolomide–HCl has been reported. The first crystallization of protonated TMZ was published in 1995 and the structure of the protonated species was ascertained based on the 15N chemical shifts.15 Later in 2013, Babu et al. reported the crystallographic data of the protonated form of TMZ and rationalized their evidence based on the calculated pKa using ChemAxon's Marvin software.16,17 The authors mentioned the poor and twinned quality of the obtained crystals and also the hydrogen atom bound to N and O, which was located in different Fourier maps.16 Thus, because of the ambiguities mentioned for the best protonation site and to seek the protonation effects on TMZ decomposition, the protonated structures with their energies accompanied by pKa values were obtained using a quantum chemical approach.
In addition, Kasende and co-workers have investigated the interaction of a water molecule with TMZ theoretically and predicted that the carbonyl O is preferred over the N atoms in the binding of a water molecule.18 However, the acidified structures were not addressed in this work and they only studied the TMZ binding site using the NBO data. In addition, it is noteworthy that, TMZ is highly unstable in an aqueous media, so only considering the favoured binding site with a water molecule without examining its degradation would be misleading.
This article aims to study the TMZ degradation mechanism under both neutral and acidic conditions to find its decomposition pathways, provide insight into the pH dependence behavior and the stability of the drug, which could be applicable for the preparation of novel pharmaceutical co-crystals of TMZ.
Methodology
Complete optimization of all structures has been carried out using density functional theory and complete basis set (CBS-4M)19 methods. DFT calculations were carried out using the B3LYP20 functional with 6-31+G(d) and 6-311++G(2d,p) basis sets for the mechanism and protonation studies, respectively. The abovementioned methods show good results in some structural-like cases.21 Furthermore, several studies examine the protonation effect at Gn(MP2) levels, and therefore, for the sake of accuracy, the G3MP2 (ref. 22) method was also selected for obtaining the protonation energies.23–25 It is good to note that the higher level CBS-QB3 method was not used instead of CBS-4M owing to its limitation to compounds with less or near 10 heavy atoms (TMZ + water has 15 heavy atoms).26
In order to confirm the exact structure of the reactants, transition states, and products, the vibrational frequencies were examined. One imaginary value was observed for all transition states; subsequently, intrinsic reactant coordinate (IRC) calculations were used to assure the presence of correct optimized transition states. The reactant and product of the IRC calculation were optimized further using the same method. Moreover, integral equation formalism variants of the polarizable continuum model (IEF-PCM)27 with UFF radii were used to study the solvent effects on the optimized structures. The other solvation model SM8 was considered to see the difference between these solvation approaches.28 This universal solvation model (SM8) has also been used for the pKa calculations. This model is known for giving very precise pKa values29 (complete procedure for obtaining pKa data is summarized in the ESI†).
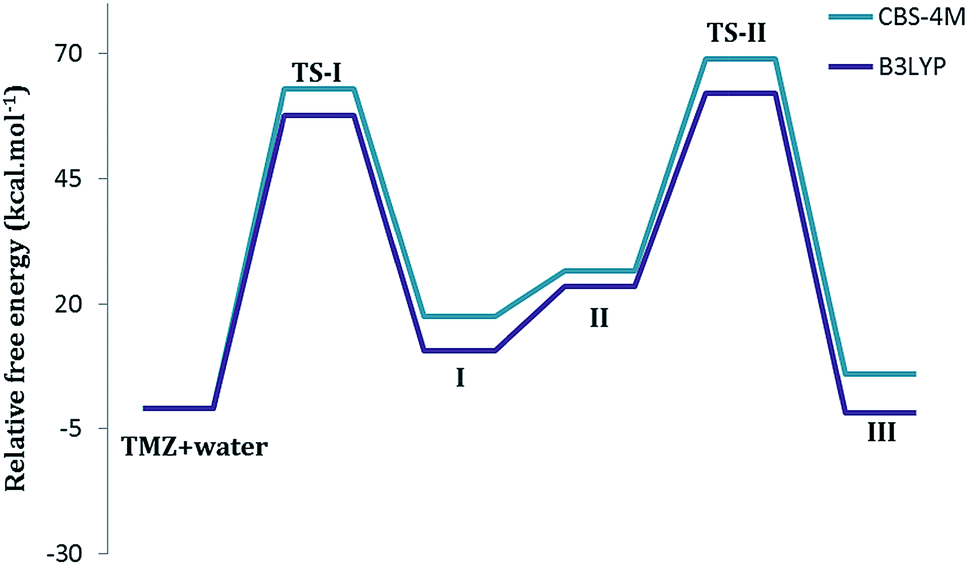
For the TMZ decomposition mechanism studies, the complex of TMZ + water (optimized as one system) was selected as the reference compound (Gref) and other energies were calculated relative to it, using the ΔG = Gref − Gx equation (Table 1). The absolute free energies (G) are also provided in the ESI.†
Table 1 The calculated relative free energies (ΔG) for both TMZ degradation paths (in kcal mol−1)
| Structurea |
Path-1 |
Path-2 |
| CBS-4M (gas) |
B3LYP/6-31+g* (gas) |
B3LYP/6-31+g* (PCM) |
B3LYP/6-31+g* (SM8) |
CBS-4M (gas) |
B3LYP/6-31+g* (gas) |
B3LYP/6-31+g* (PCM) |
B3LYP/6-31+g* (SM8) |
| The TMZ + water structure has been optimized as a reactant and all other energies were compared to this structure relatively. Separated reactants. |
| SRb |
−2.75 |
−4.15 |
−8.26 |
−7.89 |
−2.75 |
−4.15 |
−8.26 |
−7.89 |
| TS-I |
62.89 |
57.56 |
57.59 |
59.35 |
77.04 |
72.10 |
69.27 |
70.17 |
| I |
17.41 |
10.61 |
10.52 |
13.71 |
7.11 |
3.75 |
7.33 |
8.75 |
| II |
26.59 |
23.58 |
20.60 |
20.99 |
13.45 |
8.42 |
10.09 |
9.43 |
| TS-II |
68.97 |
62.17 |
63.31 |
64.30 |
52.26 |
43.16 |
48.79 |
48.14 |
| III |
5.89 |
−1.74 |
−2.89 |
3.94 |
−8.82 |
−20.93 |
−17.74 |
−10.18 |
The reactivity and electrophilicity of the TMZ species were considered using the following standard equations:
where
η,
X,
ω and
N denotes hardness, electronegativity, global electrophilicity and nucleophilicity index, respectively. The charge distribution data was achieved using the natural population analysis (NPA) approach
30 by the B3LYP/6-311++G(2d,p) method. Similarly, the nuclear independent chemical shifts (NICSs)
31 were calculated by employing the GIAO
32 method and using the B3LYP/6-311++G(2d,p) level. All the investigations were executed with Gaussian 03 and Spartan 10 software packages.
33,34
Results and discussion
Hydrolytic degradation mechanism of TMZ to MTIC
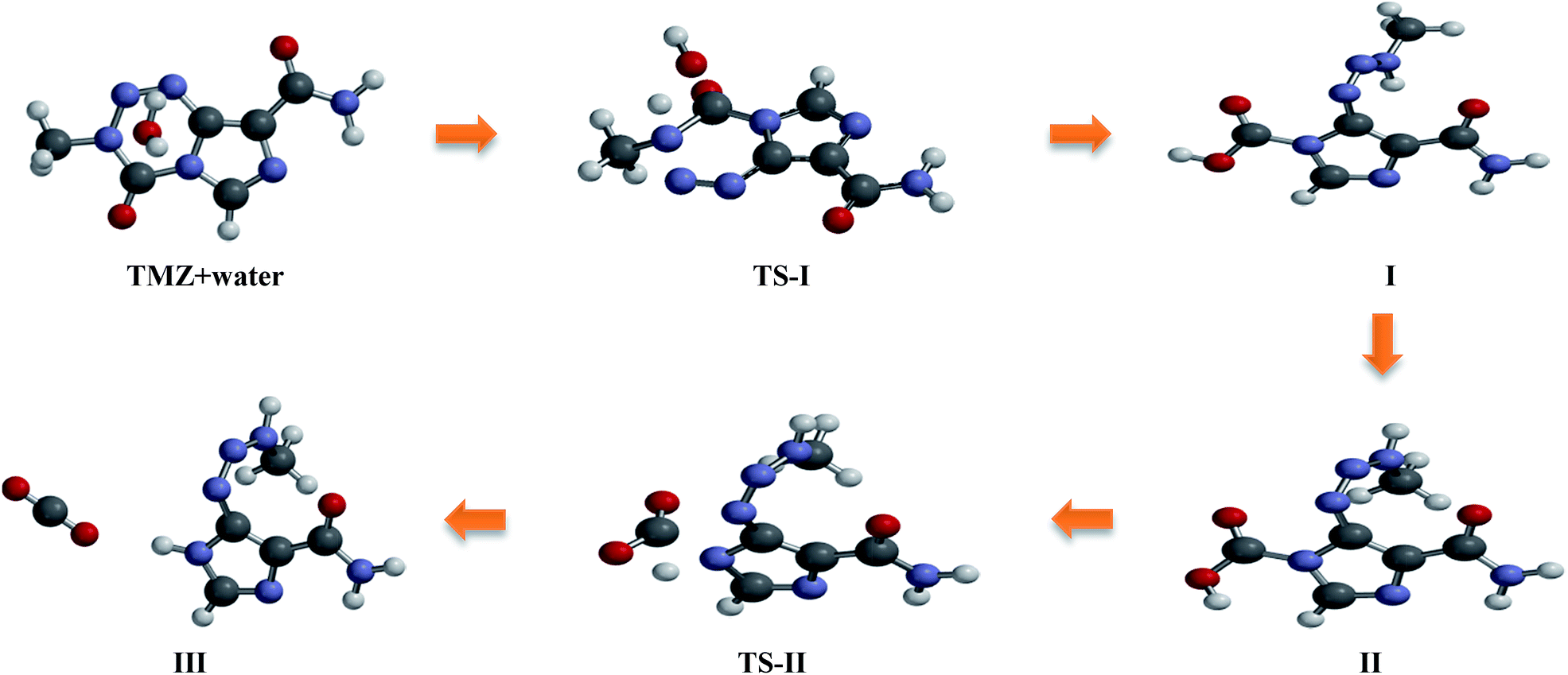
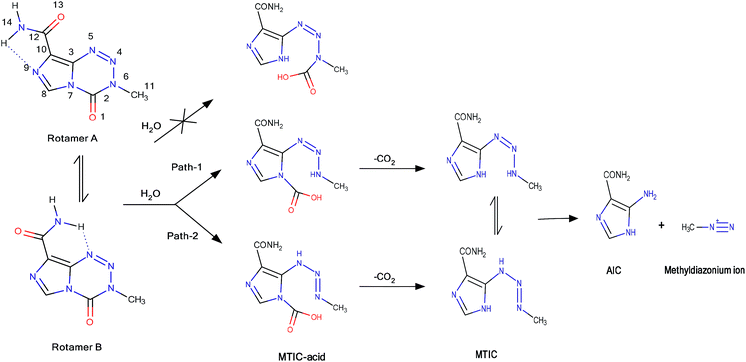
The 2D pathways of this reaction have been summarized in Scheme 1. TMZ can react with a water molecule to initiate its degradation and generate MTIC-acid. This step passes from the first transition state (TS-I in Fig. 1). There are two ways for opening the TMZ tetrazinone ring (N6–C2 and N7–C2). Although both the N6–C2 and N7–C2 bonds weaken (longer bonds) during ring-opening,35 all attempts to find the transition state for N7–C2 ring-opening using both CBS-4M and DFT levels were aborted. This may due to the electron donating effect of the methyl group, which causes the ring-opening to occur via cleavage of the N6–C2 bond. This N6–C2 bond cleavage can proceed through two different pathways (path-1 and path-2) to form MTIC-acid, which then releases CO2 molecule and generates MTIC. The MTICs produced from path-1 and path-2 are tautomeric forms, whereas the water-mediated energy barrier for this 1,3-hydrogen shift is 5.93 kcal mol−1.36 The energy diagram for both pathways reveals that in spite of the lower TS-I for path-1, the TS-II and obtained products are more energy favoured for the second path. It appears that TMZ would prevail over ring opening in the more energy favoured path-1 that forms the first MTIC tautomer but then undergoes tautomerism to generate the next MTIC tautomer (Scheme 1), which decomposes through the second path. The overall mechanism for this sequence is depicted in the ESI.†
 |
| | Scheme 1 The different pathways for the direct hydrolysis of temozolomide. | |
 |
| | Fig. 1 Path-1: TMZ decomposition. | |
Because TMZ has two rotamers, their difference is 1.35 kcal mol−1 at G3MP2 (based on the calculated free energy), and the two proposed pathways were investigated for both rotamers. The marginally increased stability of rotamer A can be explained by its aromaticity index and may be attributed to the more negative charge at N9 (−0.476) compared with N5 (−0.237). Here, the nucleus-independent chemical shifts (NICSs) were calculated for both five-membered and six-membered rings; moreover, the NICSs for the pseudo-ring of both rotamers that are the result of an intra-hydrogen bond have been estimated. Although the NICSs are almost the same for the rings, the value for rotamer A (C10–C12–N14–H⋯N9) and rotamer B (C3–C10–C12–N14–H⋯N5) pseudo-rings are −0.484 and +1.674, respectively. The negative values of NICS indicate the aromaticity and positive values denote the anti-aromaticity feature of that ring. This increased aromaticity along with the increased negative charge of N9 could justify the increased stability of rotamer A.
Note that the transition energy difference for both rotamers is roughly the same; therefore, on the basis of the Curtin–Hammett principle, the product can be derived from either rotamers and the conformational equilibrium does not alter the product distribution. Owing to this reason, only the first rotamer of TMZ has been reported in the main text and energy diagrams for the second rotamer are given in the ESI.†
Path-1 mechanism
The path-1 consists of a two-step mechanism; the first step (TS-I) is the rate determining step, which is in agreement with the experimental results obtained by Denny et al. showing that the rate-determining step must be during the early stage of the overall decomposition of TMZ.6 In this two-step pathway, one proton is transferred from the water molecule to N6 of the tetrazinone ring to produce the first transition state (the forming N–H distance is 1.18 Å and the breaking bond length (O–H) is 1.37 Å). Subsequently, the oxygen of H2O simultaneously attacks the carbonyl carbon (C2) to open the ring from the C2–N6 bond. The C2–O bond forming length is 1.89 Å and C2–N6 distance is 1.55 Å, which is longer than the C2–N6 bond (1.27 Å) in TMZ. Based on the IRC calculations, TS-I connects the complex of TMZ and H2O to MTIC-acid (Fig. 1). The gas phase relative Gibbs free energy barrier for TS-I in path-1 is 57.56 kcal mol−1 at the B3LYP level and 62.89 kcal mol−1 at the CBS-4M level (Fig. 3 and Table 1). The transition energies in aqueous media for both the PCM and SM8 solvation models have also been reported in Table 1, which shows slight change with respect to the gas data. The first transition state for rotamer B is marginally different (ESI†). In the second transition state (TS-II), one proton is transferred from OH to N7 to form a complex of MTIC and carbon dioxide. In TS-II, the N7–H forming bond length is 1.32 Å. The N7–H bond formation in TS-II is simultaneous with C2–N7 bond breaking. The C2–N7 bond length is 1.77 Å. The energy barrier for this transition is about 38.59 kcal mol−1, which when compared to the first TS is ∼20 kcal mol−1 lower in energy. The energy profile for this two-step degradation is illustrated in Fig. 3.
 |
| | Fig. 2 Path-1 relative energy profile for TMZ decomposition in vacuo. | |
Path-2 mechanism
With regard to path-1, the second path of tetrazinone ring opening also includes two steps (Fig. 2). A proton transfer from H2O to N5 of the tetrazinone ring initiates the ring opening reaction. Subsequently, the transfer of a proton to N5 leads to the formation of a N4–N6 double bond. Thereafter, the C2–N6 bond collapses as MTIC-acid is formed. This stage of the mechanism produces the first transition state (TS-I). In TS-I the N5–H bond length is 1.26 Å and the O–H distance is 1.34 Å. The N5–N4, N4–N6 and N6–C2 bond lengths in the transition state are 1.38 Å, 1.27 Å and 1.60 Å, respectively, whereas these values in intact TMZ are 1.27, 1.37 and 1.38 Angstrom, respectively. Decreasing the value of N4–N6 from 1.37 to 1.27 Å indicates the formation of a double bond. The barrier height for the first transition state is estimated to be 72.10 kcal mol−1 at the B3LYP level, which is approximately 15 kcal mol−1 more than TS-I in path-1 (Fig. 3 and 4). In Table 1, TMZ + water (treated as a system) has been chosen as the base compound and the energy of other compounds were compared with this system (absolute free energies were given in ESI†). Inclusion of water solvent keeps this energy deference at about 13 kcal mol−1. The TS-1 energy barrier for the second TMZ conformer is almost the same (ESI†). For the second step of the mechanism, the acid proton transfers to N7 of the imidazole ring to form the second transition state (TS-II). The N7–C2 bond length is 1.89 Å, which is somewhat longer than the N7–C2 bond length of path-1 (1.77 Å) second transition state. The O–H breaking bond is 1.19 Å. The first transition state for path-2, like path-1, is the rate determining step. The TS-II of path-2 has a lower energy barrier (∼5 kcal mol−1 and ∼5 kcal mol−1 in the gas and aqueous phase, respectively) in comparison to the TS-II of path-1. This indicates that path-2 is preferred over path-1 with respect to the second step of the mechanism. In addition, from a thermodynamic point of view, path-2 leads to a more stable product (Table 1).
 |
| | Fig. 3 Path-2: TMZ decomposition. | |
 |
| | Fig. 4 Path-2 relative energy profile for TMZ decomposition in vacuo. | |
Protonation of temozolomide
To probe a relation between the energy barriers of TMZ decomposition and pH, first the protonation energies were investigated for both rotamers of TMZ A and B using the multiple methods listed in Table 2. In spite of the previously reported studies, which predict the nitrogen atom in the imidazole ring (N9) as the best protonation site, the obtained values for the protonation energies demonstrate that the protonation of the oxygen atom in the TMZ amide group (O13) would be a more favourable process by 2.23 kcal mol−1 at the G3MP2 level for TMZ B (N9 protonated TMZ B is more stable than A due to the less steric hindrance and intra-hydrogen bond). Moreover, the obtained charge distribution, which has been achieved through the use of NPA, showed that more negative charge is present on O13 (−0.596). The pKa is −2.47 and −2.78 for O13 and N9, respectively (the pKa values were calculated using the procedure and method described in the ESI†). These very close values can be settled by the fact that these two protonation sites can interchange the proton between each other. The O13 and N9 protonated species may convert to each other by a relatively low barrier height (13.14 kcal mol−1) that could rationalize the simultaneous presence of both species (Fig. 5). Therefore, the pKa values have also been averaged out for both N9 and O13 at about −2.625. The negative value for the pKa clearly indicates that TMZ could only be protonated under extremely acidic conditions. In addition, as illustrated in Fig. 5, the incorporation of an explicit water molecule could decrease this barrier to 6.06 kcal mol−1. Therefore, the two protonated species can interconvert rapidly in an aqueous phase. The water molecule also decreases the energy difference between the O13w and N9w protonated structures (absolute free energies were given in the ESI†).
Table 2 Protonation free energies (ΔG) of TMZ (in kcal mol−1)
| Protonated atoma |
B3LYP/6-311++G(2d,p) |
CBS-4M |
G3MP2 |
| TMZA |
TMZB |
TMZA |
TMZB |
TMZA |
TMZB |
| Free energies of the other atoms are given in the ESI. |
| O13 |
−217.8 |
−219.4 |
−215.9 |
−217.7 |
−217.4 |
−218.7 |
| N9 |
−205.7 |
−217.2 |
−205.1 |
−216.6 |
−204.8 |
−216.4 |
| N5 |
−210.6 |
−196.1 |
−209.3 |
−195.2 |
−208.8 |
−194.2 |
| O1 |
−186.4 |
−184.0 |
−183.5 |
−180.6 |
−186.4 |
184.0 |
 |
| | Fig. 5 The gas and water-mediated interconversion between the N9 and O13 protonated forms of temozolomide. Values in bold, in parentheses, and in square brackets are the calculated relative ΔG (kcal mol−1) using the G3MP2, CBS-4M, and B3LYP/6-311++G(2d,p) methods, respectively. | |
Reactivity of TMZ in both acidic and basic condition
The reactivity of TMZ can be assessed using its relationship with electrophilicity–nucleophilicity. This concept calculates the global (ω) or local electrophilicity and nucleophilicity index (N) by employing several standard equations, which are given in the methodology section. This method for calculating reactivity has been used by several research groups.37–41
The results obtained from these equations for conformer A and B of neutral and O13 protonated TMZ with the N9 protonated species are reported in Table 3. The values of global electrophilicity reveal the extreme attitude of both neutral and protonated TMZ (strong acidic conditions) towards nucleophiles. The protonation has trivial effects on the electrophilicity of TMZ (protonation on O13 lowers the electrophilicity to some extent). In addition, the partial atomic charges supported the results in Table 3. The calculated charges (ESI†) indicate that C2 has the highest positive charge (+0.830) in TMZ, which can be attributed to the attachment of three electron-withdrawing atoms to it. Moreover, the first transition state (TS-I of path-1) in the hydrolytic degradation of TMZ on the more favourable protonated species was repeated. The N9 protonation increases the barrier of TS-I around 1 kcal mol−1. The values for protonation at O13 are 2.8 kcal mol−1 and 2.9 kcal mol−1 for O13-A and O13-B, respectively. This can also be seen qualitatively from Fig. 6, which demonstrates the LUMO orbitals of TMZ and its O13 protonated (the LUMO orbitals are considered to be attacked by the nucleophiles). The contribution of C2 in the LUMO of the O13 protonated structure is less than unprotonated TMZ (Fig. 6), so as expected, nucleophile attack at C2 is more convenient in the TMZ unprotonated structure. From all these data, it is concluded that protonation (lower pH) decreases the rate of the reaction marginally, but does not completely halt its decomposition. In addition, the extremely high protonation energy of O1, around 31 kcal mol−1 more than the O13 (Table 2), is the other factor contributing to the rate decline, which takes away the possibility for acid catalyst degradation.
Table 3 Highest occupied molecular orbital (HOMO) energy, lowest unoccupied molecular orbital (LUMO) energy, hardness (η), electronegativity (X), global electrophilicity (ω), and nucleophilicity index N. All data calculated at the B3LYP/6-311++G(2d,p) level
| Molecule |
HOMO (au) |
LUMO (au) |
η (eV) |
X (eV) |
ω (eV) |
N (eV) |
TSa |
| Gibbs free energies of the rate determining (TS-I of path-1) at the B3LYP/6-311++G(2d,p) level. |
| TMZ-A |
−0.2651 |
−0.1016 |
4.449 |
2.225 |
15.130 |
0.066 |
59.2 |
| TMZ-B |
−0.2682 |
−0.1103 |
4.296 |
2.148 |
14.613 |
0.068 |
60.4 |
| O13-A |
−0.4177 |
−0.2672 |
4.096 |
2.047 |
13.905 |
0.072 |
62.0 |
| O13–B |
−0.4170 |
−0.2670 |
4.081 |
2.040 |
13.878 |
0.072 |
63.3 |
| N9–B |
−0.4367 |
−0.2658 |
4.649 |
2.325 |
15.820 |
0.063 |
61.4 |
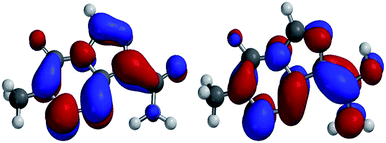 |
| | Fig. 6 The LUMO of TMZ (left) and the O13 protonated TMZ (right). | |
Finally, a brief description of basic media effects on the decomposition energies was considered using hydroxide ion in order to give a better perspective of TMZ degradation. The alkaline conditions diminish the energy barrier for degradation significantly to about 20.58 and 7.23 kcal mol−1 in TS-I (two different transition states were found). This justifies the increase in the degradation rate of TMZ from acid to basic conditions. Moreover, in order to consider the effects of other nucleophiles on TMZ decomposition, the first transition state of path-1 was determined for the nucleophiles, methylamine (MeNH2), methanol (MeOH), and acetic acid (CH3COOH). The barrier of the rate determining step (TS-I of path-1) is approximately the same for these three molecules (53–56 kcal mol−1). However, it seems that their transition state barrier is lower than that found in an aqueous media, and it has been reported that the rate of TMZ degradation to AIC reduces in media such as methanol (the rate of methanolysis is ∼2–3 days in comparison to less than one day for hydrolysis).10 In acetic acid, the optimized structure of the product prefers to retain its cyclic form and shows insignificant signs of ring opening. The product is around 40.11 kcal mol−1 less stable than the reactants (this value in an aqueous media was obtained at 17.41 kcal mol−1 (Fig. 3)). This large instability decreases the reverse transition state barrier to around 15 kcal mol−1, which could overcome the forward path significantly (55 kcal mol−1). This could be another reason for the increased stability of TMZ in acidic conditions. Nevertheless, the role of the more lipophilic acetic acid rather than water should not be ignored. The complete data for these computations are given in the ESI.†
Conclusions
The degradation reaction of the anticancer TMZ drug was simulated using quantum chemical methods. The previously reported mechanistic studies on TMZ focused more on the base-catalysed pathway; hence the neutral and acidic conditions were explored here (for comparison a basic media was also considered briefly). The results obtained demonstrate that this decomposition is a two-pathway reaction, in which the rate determining step (TS-I) for path-1 is lower than the second path. Nevertheless, it does not mean that TMZ would be decomposed through path-1 only as path-2 has a more stable TS-II and products. Thus, it has been suggested that TMZ could prevail over TS-I in considerably more favourable path-1 but then tautomerize to continue its degradation through the second path (the two paths differ in the MTIC-acid tautomeric form generated). In addition, to take the acidic effects on overall reaction into account, the protonation energies for all atoms were calculated. A novel, most stable protonated form in which O13 is protonated, was identified, which interconverts rapidly with the next preferred protonation site (as reported in previous studies). Repeated calculations on the protonated structures achieved for the rate determining step of the TMZ mechanism revealed that protonation has a decreasing effect on TMZ reactivity. However, the mechanism in neutral and near acidic mediums signifies that decomposition would occur, but at a lower rate than the more common base-catalyzed process. The parallel acid-catalyzed process did not participate in the decomposition reaction, which would equal the rate under both acidic and basic conditions, owing to the large protonation energy for O1. In addition, the electrophilicity and nucleophilicity index were investigated to support these consequences.
Acknowledgements
The authors gratefully acknowledge the Medical Biology Research Center, Kermanshah University of Medical Sciences, Kermanshah, Iran, and the Research and Computational Lab of Theoretical Chemistry and Nano Structures of Razi University Kermanshah-Iran for supporting this study.
Notes and references
- E. S. Newlands, G. Blackledge, J. A. Slack, C. Goddard, C. J. Brindley, L. Holden and M. F. Stevens, Cancer Treat. Rep., 1985, 69, 801 CAS.
- H. S. Friedman, T. Kerby and H. Calvert, Clin. Cancer Res., 2000, 6, 2585 CAS.
- M. J. van den Bent, M. J. Taphoorn and A. A. Brandes, J. Clin. Oncol., 2003, 21, 2525 CrossRef CAS.
- I. Quirbt, S. Verma, T. Petrella, K. Bak and M. Charette, Curr. Oncol., 2007, 14, 27 CrossRef CAS.
- W. J. Hwu, Oncology, 2000, 14, 25 CAS.
- B. J. Denny, R. T. Wheelhouse, M. F. G. Stevens, L. L. H. Tsang and J. A. Slack, Biochemistry, 1994, 33, 9045 CrossRef CAS.
- R. T. Wheelhouse and M. F. G. Stevens, J. Chem. Soc., Chem. Commun., 1993, 1177 RSC.
- H. Kim, P. Likhari, D. Parker, P. Statkevich, A. Marco, C. Lin and A. A. Nomeir, J. Pharm. Biomed. Anal., 2001, 24, 461 CrossRef CAS.
- O. Braverman, R. Feishtein, A. Weisman and J. Kaspi, US Pat., 0222792 A1, Oct. 05, 2006.
- N. J. Babu, P. Sanphui and A. Nangia, Chem.–Asian J., 2012, 7, 2274 CrossRef CAS PubMed.
- N. J. Babu, L. S. Reddy, S. Aitipamula and A. Nangia, Chem.–Asian J., 2008, 3, 1122 CrossRef CAS PubMed.
- P. Sanphui, N. J. Babu and A. Nangia, Cryst. Growth Des., 2013, 13, 2208 CAS.
- A. Nangia, N. J. Babu and P. Sanphui, WO Pat., 036676 A2, Mar. 31, 2011.
- I. Adin and C. Iustain, US Pat., 0187206 A1, Aug. 25, 2005.
- R. T. Wheelhouse, D. E. V. Wilman, W. Thomson and M. F. G. Stevensa, J. Chem. Soc., Perkin Trans. 1, 1995, 249 RSC.
- N. J. Babu, P. Sanphui, N. K. Nath, U. V. R. Khandavilli and A. Nangia, CrystEngComm, 2013, 15, 666 RSC.
- pKa calculator, Marvin 5.10.1, ChemAxon, 2012, http://www.chemaxon.com Search PubMed.
- O. E. Kasende, A. Matondo, M. Muzomwe, J. T. Muya and S. Scheiner, Comput. Theor. Chem., 2014, 1034, 26 CrossRef CAS PubMed.
- G. A. Petersson, J. A. Montgomery, M. J. Frisch and J. W. Ochterski, J. Chem. Phys., 2000, 112, 6532 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- D. L. Cao, F. D. Ren, J. L. Wang and W. L. Wang, J. Mol. Struct.: THEOCHEM, 2007, 805, 53 CrossRef CAS PubMed.
- L. A. Curtiss, P. C. Redfern, K. Raghavachari, V. Rassolov and J. A. Pople, J. Chem. Phys., 1999, 110, 4703 CrossRef CAS PubMed.
- P. J. Amorim Madeira, P. D. Vaz, R. J. N. Bettencourt da Silva and M. H. Florêncio, ChemPlusChem, 2013, 78, 1149 CrossRef CAS PubMed.
- S. Gronert, D. C. Simpson and K. M. Conner, J. Am. Soc. Mass Spectrom., 2009, 20, 2116 CrossRef CAS PubMed.
- P. V. Bharatam, D. S. Patel and P. Iqbal, J. Med. Chem., 2005, 48, 7615 CrossRef CAS PubMed.
- G. A. Petersson, J. A. Montgomery Jr, M. J. Frisch and J. W. Ochterski, J. Chem. Phys., 2000, 112, 6532 CrossRef PubMed.
- M. T. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef PubMed.
- A. V. Marenich, R. M. Olson, C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2007, 3, 2011 CrossRef CAS.
- S. Gangarapu, A. T. M. Marcelis and H. Zuilhof, ChemPhysChem, 2013, 14, 990 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. J. Weinhold, Chem. Phys., 1985, 83, 735 CAS.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld and H. Jiao, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- F. London, J. Phys. Radium, 1937, 8, 397 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven Jr, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian-03 suite of programs, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- Spartan 10 Package, Windows Version, Wavefunction Inc., USA, 2011 Search PubMed.
- P. R. Lowe, C. E. Sansom, C. H. Schwalbe, M. F. G. Stevens and A. S. Clark, J. Med. Chem., 1992, 35, 3377 CrossRef CAS.
- K. G. Doucet, J. F. Glister and C. C. Pye, Can. J. Chem., 2010, 8, 709 CrossRef PubMed.
- P. A. Krieter, A. E. Colletti, G. A. Doss and R. R. Miller, Drug Metab. Dispos., 1994, 22, 625 CAS.
-
(a) H. Toufar, K. Nulens, G. O. A. Janssens, W. J. Mortier, R. A. Schoonheydt, F. De Proft and P. Geerlings, J. Phys. Chem. B, 1996, 100, 15383 CrossRef CAS;
(b) F. De Proft and P. Geerlings, Chem. Rev., 2001, 101, 1451 CrossRef CAS PubMed;
(c) P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed;
(d) T. Fievez, N. Sablon, F. DeProft, P. W. Ayers and P. J. Geerlings, J. Chem. Theory Comput., 2008, 4, 1065 CrossRef CAS;
(e) G. Roos, P. Geerlings and J. Messens, J. Phys. Chem. B, 2009, 113, 13465 CrossRef CAS PubMed;
(f) J. T. Muya, F. De Proft, P. Geerlings, M. T. Nguyen and A. Ceulemans, J. Phys. Chem. A, 2011, 115, 9069 CrossRef CAS PubMed.
-
(a) N. T. Brown and N. Mora-Diez, J. Phys. Chem. B, 2006, 110, 9270 CrossRef PubMed;
(b) L. A. M. M. Barbosa and R. A. van Santen, J. Mol. Struct.: THEOCHEM, 2000, 497, 173 CrossRef CAS.
- P. K. Chattaraj, U. Sarkar and D. R. Roy, Chem. Rev., 2006, 106, 2065 CrossRef CAS PubMed.
- P. K. Chattaraj, A. Chakraborty and S. Giri, J. Phys. Chem. A, 2009, 113, 10068 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The energy profile for rotamer B of TMZ as well as charge distribution and all atoms protonation energies. See DOI: 10.1039/c5ra04680g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.