DOI:
10.1039/C5RA04643B
(Paper)
RSC Adv., 2015,
5, 41445-41456
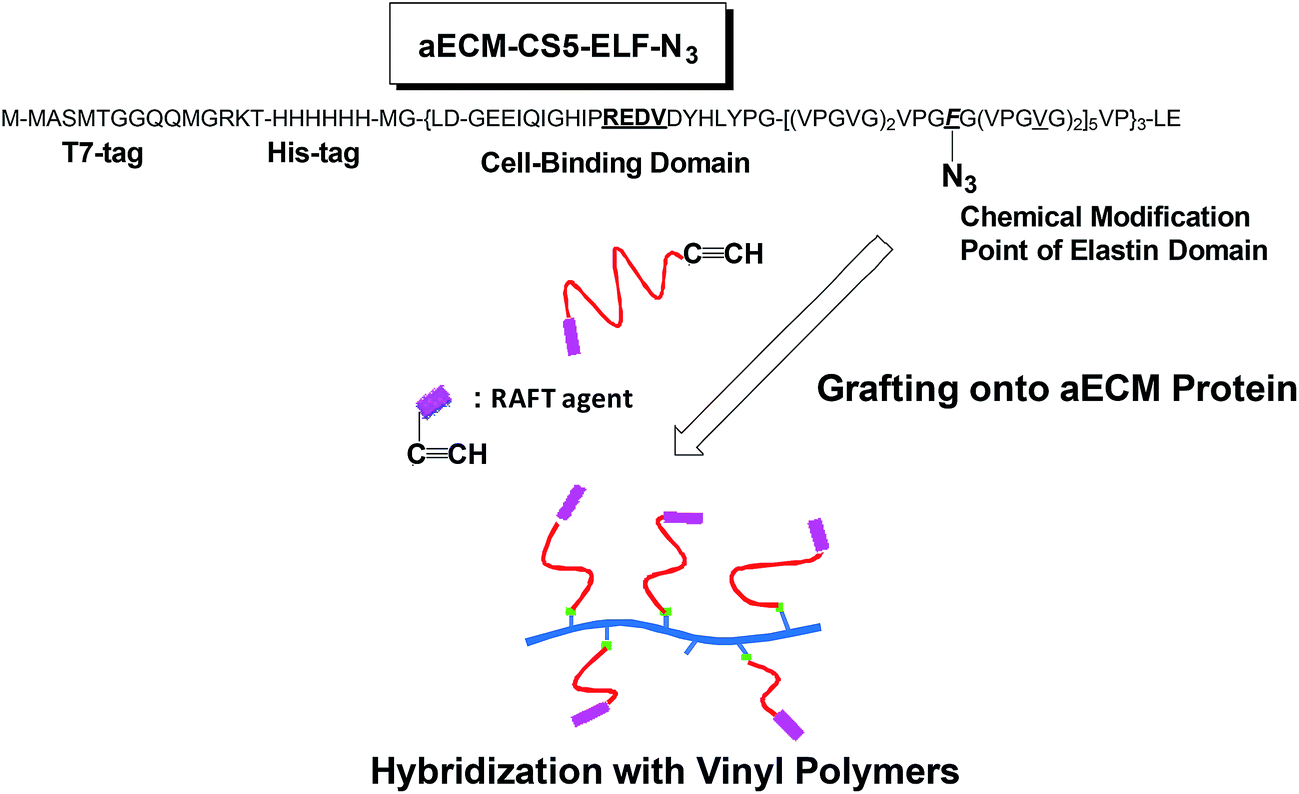
Click grafting of alkyne-containing vinyl polymers onto biosynthesized extracellular matrix protein containing azide functionality and adhesion control of human umbilical vein endothelial cells†
Received
16th March 2015
, Accepted 30th April 2015
First published on 30th April 2015
Abstract
In vivo incorporation of a phenylalanine (Phe) analogue, p-azidophenylalanine (p-N3Phe) into an artificial extracellular matrix protein (aECM-CS5-ELF) was accomplished using a bacterial expression host that harbors the mutant phenylalanyl-tRNA synthetase (PheRS) with an enlarged binding pocket, in which the Ala294Gly/Thr251Gly mutant PheRS (PheRS**) was expressed under the control of T7 promoters. In this study, biosynthesized aECM-CS5-ELF containing p-N3Phe (aECM-CS5-ELF-N3) was coupled with alkyne-containing vinyl polymers prepared via controlled radical polymerization of three vinyl monomers, (styrene, acrylamide, and N-isopropylacrylamide) using a trithiocarbonate as the RAFT agent. Grafting of the vinyl polymers onto the aECM was accomplished via a copper-catalyzed alkyne–azide click reaction. The lower critical transition temperature (LCST) was evaluated, as well as the solubility in aqueous and organic media, which are dependent on the incorporation ratio of p-N3Phe and species of graft chains, in which the LCST behavior was altered remarkably when poly(N-isopropylacrylamide) moieties were attached as side chains. Circular dichroism measurements indicate conformational change was not induced by the grafting. Specific adhesion of human umbilical vein endothelial cells (HUVECs) onto the (aECM-CS5-ELF-N3)-graft-poly(N-isopropylacrylamide) composite surface and subsequent temperature-sensitive detachment were also demonstrated.
Introduction
Tissue engineering has become a key therapeutic tool in the treatment of damaged or diseased organs and tissues, such as blood vessels (small-diameter vascular graft) and urinary bladders.1,2 The extracellular matrix (ECM), which includes protein ligands, such as elastin and fibronectin, is an insoluble macromolecular complex that forms an environmental substrate outside the cells of organs and tissues.3–5 The cell-to-ECM contact plays an important role in the regulation of biological processes such as wound healing, organogenesis, and metastasis.4,5 Tirrell et al. have reported the results of studies on artificially designed analogues of ECM proteins (aECMs) containing an elastin sequence and the fibronectin cell binding domain (CS5), aECM-CS5-ELF.6 They also examined how these aECMs affect the adhesive behavior of human umbilical vein endothelial cells (HUVECs).6 The elastin-like sequences give these materials elasticity and mechanical integrity,6b,6d while cell adhesion and cell spreading on fibronectin are maintained. Either when fibronectin coats a plate or is present in the extracellular matrix, interaction with HUVEC are mediated by integrin receptors such as α5β1,7 and α4β1.8,11 The sequence motif REDV, found within the CS5 domain, is thought to be the shortest sequence capable of α4β1 integrin binding.8b In Tirrell's studies on aECM-CS5-ELF,6c,6e HUVEC adhesion and spreading were dependent of the presence of the CS5 domain of fibronectin. Nonetheless, major challenges still need to be overcome, in particular, the construction of tissues with high cell densities and the prevention of post-transplant inflammation. Furthermore, the poor solubility and processability of aECM-CS5-ELF is inhibiting the expansion of the range of further applications, and must be addressed.
When designing new advanced biomaterials to be used in tissue engineering, facile chemical modification of cell–matrix interactions is essential. One approach is incorporation of non-natural amino acids containing a reactive functional group9 into an aECM for post-translational chemical modification while maintaining the structural integrity of the protein. For example, incorporation of phenylalanine (Phe) analogues [including p-N3Phe, p-bromophenylalanine (p-BrPhe), and p-iodophenylalanine (p-IPhe) having an orthogonal functional group] into a recombinant protein enables chemical modification via “click” reactions, Heck reactions, or Sonogashira couplings, and has been accomplished using a bacterial expression host that harbors a mutated phenylalanyl-tRNA synthetase (PheRS) with an enlarged binding pocket.10,11 Tirrell et al. previously identified mutations which enlarge the Phe binding cavity of E. coli PheRS, Ala294Gly10a and Thr251Gly,10b and produced the corresponding Ala294Gly (PheRS*)10a and Thr251Gly/Ala294Gly10b (PheRS**) mutants, respectively. The Thr251Gly/Ala294Gly double mutant, PheRS**, effectively incorporates even p-acetylphenylalanine (p-AcPhe) into mouse dihydrofolate reductase when the T5 system is used.10b Although the amount of material produced is insufficient due to the moderate strength of the T5 promoter, we previously reported that this limitation can be overcome using a combination of T7 promoter and T7 RNA polymerase.11 The T7 RNA polymerase has greater activity and is more selective than the endogenous E. coli RNA polymerase.12–14 The T7 expression system (pET system) is capable of producing large amounts of protein (up to 100 mg L−1 of culture medium).11 We also reported PEGylation of aECM using a click strategy, and showed that the increased hydrophilicity upon PEGylation of the elastin domain results in lowered non-specific adhesion of human umbilical vein endothelial cells (HUVECs).11 We hypothesize that in order to control the hydrophilicity of the elastin-type sequences within the aECM by conjugation of the vinyl polymers, poly(acrylamide) (PAAm) and poly(N-isopropylacrylamide) (PNIPAM), further advanced biomaterials should be provided. Particularly, these new materials can be utilized in a promising tissue engineering approach relying on the use of cell culture surfaces grafted to PNIPAM, which is based on the idea that cell adhesion/detachment on PNIPAM-modified substrates can be achieved via a simple temperature switch, the LCST.15
Recently, chemical modification of several proteins via grafting from procedure has been studied using the reversible addition fragmentation chain transfer (RAFT) radical polymerization technique.16 However, until now, few reports dealing with grafting onto procedure was reported, as far as we know.17 In the present study, biosynthesized aECM-CS5-ELF containing p-N3Phe (aECM-CS5-ELF-N3) was prepared using PheRS** and a T7 promoter.11 Next, alkyne-containing trithiocarbonate was generated for use as the RAFT agent for controlled radical polymerization of three vinyl monomers: styrene, acrylamide, and N-isopropylacrylamide. Hybridization of the aECM with vinyl polymers is accomplished via a copper-catalyzed alkyne–azide click reaction18 based on a “grafting onto” procedure (Fig. 1). The lower critical transition temperature (LCST) was evaluated, as well as the solubility in aqueous and organic media, which is dependent on the incorporation ratio of p-N3Phe as well as the length and type of graft chains. Notably, the LCST behavior was altered significantly when PNIPAM was attached as the side chains. From the circular dichroism measurements, a conformational change (ex. from random coil to α-helix) was investigated before and after the grafting. Specific adhesion of HUVECs onto the aECM-CS5-ELF/vinyl polymers composite surface was also demonstrated.
 |
| | Fig. 1 Incorporation of p-azido-L-phenylalanine into the aECM-CS5-ELF-F protein and coupling with vinyl polymers prepared via RAFT radical polymerization of vinyl monomers. | |
Experimental section
Materials
p-N3Phe was obtained from Bachem AG (Hauptstrasse 114/4416 Bubendorf, Switzerland). The twenty natural amino acids and 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) were obtained from Aldrich. The cell culture medium and trypsin/EDTA solution were obtained from Lonza. Crystal violet was obtained from Wako. The GREDVY peptide was prepared via Fmoc solid-phase peptide synthesis in our laboratory and characterized by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy (H2N-GREDVY-COOH + Na+: m/z = 762.2).
Measurements
The 1H NMR spectra of the aECMs were acquired at 27 °C using a Bruker DPX200 spectrometer (200 MHz) or a Bruker DPX-600 (600 MHz) spectrometer (Bruker Japan, Japan). The number-average molecular weight (Mn) and polydispersity index (Mw/Mn) of the polymers were estimated by size-exclusion chromatography (SEC) using a Tosoh DP8020 pump system, a refractive index (RI) detector (Tosoh RI-8020), and a TSKgel SuperMultiporeHZ-M column (eluent, chloroform; flow rate, 0.35 mL min−1; temperature, 40 °C; Tosoh Corp.) using poly(styrene)s as the calibration standard. MALDI-TOF mass spectra were measured with a Voyager RN using dithranol as a matrix reagent. NaI was used as the cationization salt to generate sodium-cationized ions ([M + Na]+). The purity and molecular weight of the purified aECM-CS5-ELF-N3 were confirmed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). FT IR spectra (KBr disc) were acquired using a JASCO FTIR-430 spectrometer.
Preparation of alkyne-containing RAFT Agent
Following the procedure reported by Brittain et al.,19 DDMAT (0.5 g, 1.37 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 0.39 g, 2.0 mmol), 4-dimethylaminopyridine (DMAP, 0.25 g, 2.0 mmol), and 5 mL of dichloromethane were added to a round-bottomed flask and stirred for a few minutes under a nitrogen atmosphere (Scheme 1). Into the mixture, 0.31 mL (4.11 mmol) of 3-butyn-1-ol was added, and stirred overnight at room temperature. After diluting 30 mL of dichloromethane in a round-bottomed flask, the product was washed with 30 mL of aqueous HCl (0.1 M), 30 mL of water, and 30 mL of 10% aqueous NaCl three times, and dried under reduced pressure. The expected product 1 was obtained as a yellow syrup (0.49 g, 85% yield) without further purification. 1H NMR (200 MHz, CDCl3, δ, ppm): 0.87 (3H, t, CH2CH3), 1.2–1.4 (20H, m, CH2CH2), 1.65 (6H, s, C(CH3)2), 1.97 (1H, t, HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 2.53 (2H, dt, CH2CC), 3.28 (2H, t, –CH2S), 4.2 (2H, t, OCH2C).
C), 2.53 (2H, dt, CH2CC), 3.28 (2H, t, –CH2S), 4.2 (2H, t, OCH2C).
 |
| | Scheme 1 Preparation of alkyne-containing RAFT agent 1. | |
Plasmid construction
The incorporated polylinker encodes a T7-tag, a hexahistidine tag (His tag), and an enterokinase cleavage site (Fig. 1). This linker was cloned into a pET28 plasmid between the Nco I and Xho I sites. The pET28-CS5-ELF-PheRS* plasmid10c was used as a template for a polymerase chain reaction (PCR) mutagenesis to generate the coding sequence for PheRS**. This new plasmid was designated pET28-CS5-ELF-PheRS**.11
Incorporation of p-N3Phe into the aECM-CS5-ELF
Buffer and media were prepared according to standard protocols. A phenylalanine auxotrophic derivative of E. coli BL21(DE3), constructed in our laboratory and designated AF [HsdS gal (ncIts857 ind 1 Sam7 nin5 lacUV5-T7 gene 1) pheA], was used as the expression host. The AF strain was transformed with the repressor plasmid pLysS-IQ and with pET28-CS5-ELF-PheRS** to afford the expression strain AF-IQ[pET28-CS5-ELF-PheRS**]. We have already reported the in vivo translational activities of the Phe analogues, (p-N3Phe, p-BrPhe, p-IPhe, p-ethynylphenylalanine, p-cyanophenylalanine, and p-AcPhe).11 M9 minimal medium (1 L) supplemented with 0.4% glucose, 35 mg L−1 thiamine, 0.1 mM MgSO4, 0.1 mM CaCl2, the 20 naturally occurring amino acids (at 40 mg L−1 each), and antibiotics (kanamycin 25 mg L−1, chloramphenicol 20 mg L−1) was inoculated with 300 mL of culture containing the expression strain. When the optical density at 600 nm reached 0.7–1.0, a medium shift was performed. The cells were sedimented via centrifugation for 7 min at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 × g and 4 °C, and the supernatant liquid was removed. The cells were resuspended in supplemented M9 medium deficient in Phe and Trp. Protein expression was induced after the medium shift using isopropyl-β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Ten minutes later, a Phe analogue was added at a concentration of 250 mg L−1.11 The cells were cultured for four hours post-induction, and protein expression was monitored via SDS-polyacrylamide gel electrophoresis (PAGE) using a normalized OD600 of 0.5 per sample.
100 × g and 4 °C, and the supernatant liquid was removed. The cells were resuspended in supplemented M9 medium deficient in Phe and Trp. Protein expression was induced after the medium shift using isopropyl-β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Ten minutes later, a Phe analogue was added at a concentration of 250 mg L−1.11 The cells were cultured for four hours post-induction, and protein expression was monitored via SDS-polyacrylamide gel electrophoresis (PAGE) using a normalized OD600 of 0.5 per sample.
Protein purification (for 1 L of culture)
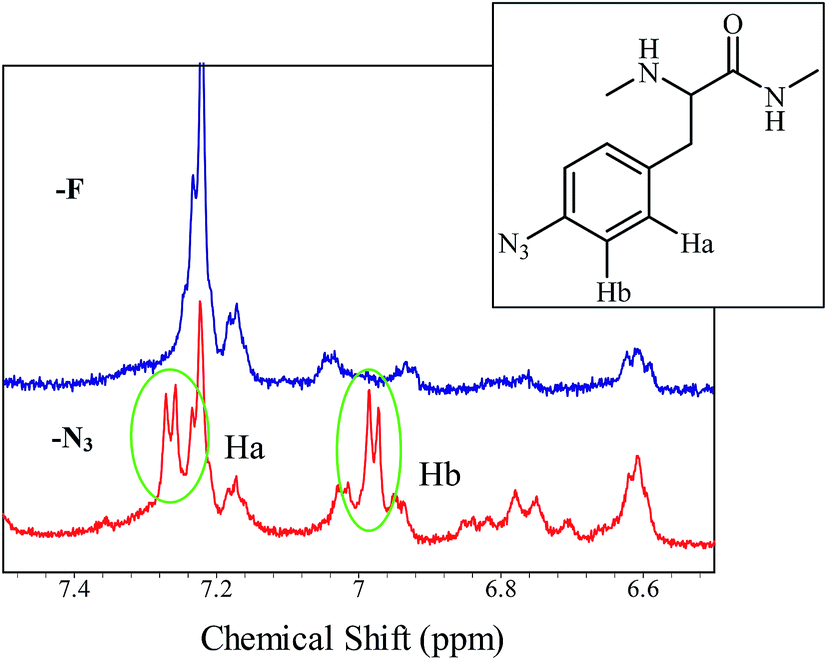
The purification scheme, which takes advantage of the inverse temperature transitions of aECMs, has been reported elsewhere.6b,11 Briefly, the wet cell mass was dispersed in TEN buffer at a concentration of 1 g mL−1, frozen, and defrosted at 4 °C with addition of 1.0 μg mL−1 DNase, 10 ng mL−1 RNase, and 50 ng mL−1 phenylmethylsulfonyl fluoride (PMSF). The biosynthesized aECMs, aECM-CS5-ELF-N3s, which were partitioned into the pellet of the whole-cell lysate after centrifugation (20100 × g, 60 min, 37 °C), were resuspended in 4 M urea. The solutions were centrifuged (20100 × g, 60 min, 4 °C) to remove non-protein cellular debris and then dialyzed against water (3 days, 4 °C) using cellulose tubing (cut off 14000). The precipitates were removed via centrifugation (20100 × g, 60 min, 2 °C), and the clarified supernatant were then lyophilized. The purity of the proteins and the uniformity of their molecular weights were confirmed via SDS-PAGE. The relative amounts of p-N3Phe analogues incorporated were derived from the areas of the 600 MHz 1H NMR spectra aromatic peaks (Fig. 2).
 |
| | Fig. 2 Expanded 1H NMR spectra (in DMSO-d6 at ambient temperature, 600 MHz) of purified aECM-CS5-ELF-N3 using PheRS** (red line) as the mutated phenylalanyl-tRNA synthetases and aECM-CS5-ELF-F (blue line). | |
RAFT radical polymerization of styrene using RAFT agent 1
Following the reported procedure,19 styrene (1.385 g, 13 mmol), RAFT agent 1 (125 mg, 0.3 mmol), α,α′-azoisobutyronitrile (AIBN, 3 mg, 0.018 mmol) and 1.5 mL of toluene were added to a round-bottom flask. After degassing, the reaction mixture was stirred at 90 °C for 18 hours under an inert atmosphere. The reaction was stopped by quenching in an ice bath. Polystyrene (PSt) was obtained after precipitation in cold methanol. SEC was used to calculate molecular weight (Mn = 1700 g mol−1) and polydispersity (1.2). (0.55 g, 36% yield). 1H NMR (200 MHz, CDCl3, δ, ppm): 0.88 (ω-terminus, t, CH2CH3), 1.26 (ω-terminus, m, CH2), 1.5 (α-terminus, s, C(CH3)2), 1.3–1.7 (2H, br, CH2CH), 1.7–2.0 (1H, br, CH2CH), 2.20 (α-terminus, s, CH2CCH), 3.25 (ω-terminus, t, –CH2SC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), 6.31–6.87 (br, aromatic protons), 6.89–7.23 (br, aromatic protons).
S), 6.31–6.87 (br, aromatic protons), 6.89–7.23 (br, aromatic protons).
RAFT radical polymerization of acrylamide using RAFT agent 1
Following the reported procedure,19 RAFT agent 1 (10 mg, 0.024 mmol), acrylamide (100 mg, 1.4 mmol), 1 mL of dimethylsulfoxide (DMSO), and AIBN (0.13 mg, 0.014 mmol) were combined in a Schlenk flask. After degassing, the reaction mixture was heated to 70 °C for 2 days under N2. The polymerization was quenched by cooling the flask in an ice bath, and PAAm was obtained after precipitation in cold methanol. The molecular weight of PAAm was calculated via 1H NMR spectroscopy (91 mg, 82% yield, Mn = 3200 g mol−1). 1H NMR (200 MHz, D2O, δ, ppm): 0.57 (ω-terminus, t, CH2CH3), 0.76, 1.14 (ω-terminus, br, CH2CH2), 1.62 (2H, br, CH2CH), 2.15 (1H, br, CH2CH), 2.53 (α-terminus, br, CH2CC), 2.86 (ω-terminus, t, –CH2S), 4.11 (α-terminus, br, COOCH2).
RAFT radical polymerization of N-isopropyl acrylamide using RAFT agent 1
RAFT agent 1, (8.3 mg, 0.02 mmol), N-isopropylacrylamide (56.6 mg, 0.5 mmol), 1 mL of tetrahydrofuran (THF), and AIBN (1.6 mg, 0.01 mmol) were mixed in a Schlenk flask. After degassing, the reaction mixture was heated to 60 °C for 2 days under N2. The polymerization was quenched by cooling the flask in an ice bath, and PNIPAM was obtained (65 mg, 24% yield) after precipitation in n-hexane. SEC was used to calculate molecular weight (Mn = 4800 g mol−1) and polydispersity (1.6). 1H NMR (200 MHz, CDCl3, δ, ppm): 0.88 (ω-terminus, t, CH2CH3), 1.14 (6H, br, NHCH(CH3)2), 1.25 (ω-terminus, m, CH2), 1.65 (2H, br, CH2CH), 2.09 (1H, br, CH2CH), 2.54 (α-terminus, br, CH2CC), 3.33 (ω-terminus, t, –CH2SCS), 4.01 (1H, br, NCH), 4.16 (α-terminus, br, COOCH2), 5.78–6.94 (br, NH).
Preparation of tris-(3-hydroxypropyltriazolylmethyl)amine (THPTA, ligand for click reaction)
Synthesis of 3-azido-1-propanol. Sodium azide (1.43 g, 22.0 mmol) was added to a solution of 3-bromo-1-propanol (1.54 g, 11.1 mmol) in DMF (6 mL) and stirred at room temperature for 48 hours. The mixture was diluted with 30 mL of CH2Cl2 and washed 5 times with 30 mL of 10% aqueous NaCl. The organic layer was dried, filtered, and concentrated to give the product. Yield: 57%. 1H NMR (CDCl3) δ (ppm): 3.73 (t, 2H, 5.6 Hz), 3.45 (t, 2H, 6.8 Hz), 2.55 (br, 1H), 1.83 (m, 2H).
Synthesis of THPTA. Tripropargylamine (0.13 g, 1 mmol) in an acetonitrile/methanol solution (3 mL) was treated sequentially with 3-azido-1-propanol (0.4 g, 4 mmol), 2,6-lutidine (0.11 g, 1 mmol), and Cu(I)Br (14.3 mg, 1 × 10−2 mmol). Upon addition of the copper salt, the reaction was cooled in an ice bath. After the mixture was stirred at room temperature for 3 days, the reaction mixture was evaporated under reduced pressure and dissolved in methanol. The crude product was precipitated in acetonitrile. Yield: 71%. 1H NMR (DMSO-d6) δ (ppm): 8.03 (s, 3H), 4.66 (t, 3H, 4.9 Hz), 4.44 (t, 6H, 6.8 Hz), 3.62 (s, 6H), 3.40 (q, 6H, 5.4 Hz), 1.97 (m, 6H) (see also Fig. S1†).
Grafting of poly(acrylamide) onto aECM-CS5-ELF
aECM-CS5-ELF-N3 [10 mg, 2.5 × 10−4 (azido unit) mmol], PAAm (12 mg, 3.8 × 10−3 mmol), THPTA (14.3 mg, 3.3 × 10−2 mmol), Cu(I)Br (1.43 mg, 1 × 10−3 mmol) and 4 mL of phosphate buffer (pH 7.4) were added to a round-bottom flask and stirred at room temperature for 4 days under an inert atmosphere. The aqueous solution was dialyzed water (3 days, 4 °C) and lyophilized. IR (KBr) cm−1: 2964 (νC–H), 1665 [νCO (amideI)], 1542 [δNH (amideII)], 1455, 1418 (δC–H).
LCST measurements
The LCST values were measured using aECM aqueous solutions (1 mg/1 mL). The temperature was increased at a rate of 1 °C min−1 while the percent transmission at 350 nm of the solution was measured using a V-550 spectrometer (JASCO, Tokyo, Japan).
Coating of aECM-CS5-ELF-N3 having PAAm and PNIPAM side chains onto a commercially-available cultivation dish
Physical mixtures of aECM-CS5-ELF-N3/(aECM-CS5-ELF-N3)-graft-PAAm (2 mg/2 mg) and aECM-CS5-ELF-N3/(aECM-CS5-ELF-N3)-graft-PNIPAM (2 mg/2 mg) were dissolved in water (1 mL), respectively. Small amounts of the solutions (80 μL) were dropped and spread onto 40 mm × 15 mm Petri dishes. The dishes were dried at 40 °C for 4 h. The dried dishes were irradiated using an ASAHI SPECTRA MAX-303 (ASAHI SPECTRA, Tokyo, Japan) (λ = 250–400 nm) for 30 min to induce cross-linking via nitrene chemistry of azido group. The dishes were immersed in cold 8 M urea buffer for 3 min and rinsed with 10% aqueous NaCl. Then, they were immersed in ethanol and dried at 40 °C for 1 hour.
Cell culture
The HUVECs (Lonza Walkersville, Inc.) were maintained in a 37 °C, 5% CO2, humidified environmental chamber. The cells were grown in endothelial growth medium-2 (EGM-2, 2% FBS) (Lonza Walkersville), which was replaced every 2 days. Near confluent HUVEC cultures were passaged by treatment with 0.025% trypsin/0.01% EDTA (Lonza Walkersville).
Cell spreading
HUVECs in EGM-2 were added to 40 mm × 15 mm Petri dishes at a concentration of 5000 cells per Petri dish. Petri dish was made of polystyrene, we used after washing with ethanol. After 3 days, the plates were removed from the environmental chamber and the cells were imaged using a 10× phase contrast objective on a Nikon Eclipse TS100/TS100-F epifluorescence microscope (Tokyo, Japan).
Cell resistance to detachment
HUVECs incubated for 3 days in EGM-2 were added to a Petri dish. A solution of the GREDVY peptide in phosphate buffered saline (PBS) (1.5 mM) was added. After 30 min of incubation at 37 °C and 5% CO2, non-adhered cells were removed by inversion of the plate and rinsing with PBS. The cells were stained with crystal violet, and thoroughly rinsed with water. The dye was solubilized using a 1% SDS solution. The absorbance was measured at 595 nm with a Jasco J820K spectropolarimeter. At least three independent experiments were carried out in order to verify reproducibility.
Results and discussion
Incorporation of pN3Phe into aECM-CS5-ELF-F
Incorporation of p-N3Phe into a protein can be used to fabricate a chemoselective access point for alkyne-initiated vinyl polymers prepared via RAFT radical polymerization. For this work, the phenylalanine auxotrophic E. coli strain (AF) carrying the repressor plasmid pLysS-IQ, AF-IQ, was transformed with pET-CS5-ELF-PheRS** to generate the expression system AF-IQ[pET-CS5-ELF-PheRS**]. Expression of aECM-CS5-ELF-N3 was under the control of an inducible T7 promoter. The expression system was induced with IPTG and cultivated for an additional four hours to allow for the biosynthesis of aECM-CS5-ELF-N3. The whole cell lysates and purified aECM proteins were analyzed via SDS-PAGE, and the gels show a significant amount of aECM protein with a calculated molecular weight of 42.6 kDa, the expected aECM proteins were biosynthesized. Using PheRS**, p-N3Phe (250 mg L−1 culture) was readily incorporated into aECM-CS5-ELF, and in most cases produced more than 50 mg of the target proteins from 1 L culture.11 Analysis of the 1H NMR spectrum of aECM-CS5-ELF-N3 shows that the incorporation level of pN3Phe was 67% when PheRS** was used (Fig. 2, red line). This incorporation level is derived from the ratio of the area of the aromatic signal at 7.20 ppm ascribed to Phe and the signal at 6.98 ppm (d, 8.1 Hz) ascribed to p-N3Phe.
Alkyne-containing vinyl polymers prepared via RAFT radical polymerization technique
DDMAT was esterified using 3-butyn-1-ol to afford the expected alkyne-containing trithiocarbonate RAFT agent 1 for use in the controlled radical polymerization of vinyl monomers (Scheme 1). Trithiocarbamates are also known to be effective RAFT agents for mediating the polymerization of conjugated vinyl monomers.16,17 The results of the RAFT radical polymerizations of styrene are summarized in Table 1. The trithiocarbamate 1 seemed to induce reversible chain transfer reactions. When styrene was used as the vinyl monomer, the prepared trithiocarbamate 1, was effective for the control of molecular weight (runs 1–3). In run 1, the molecular weight (Mn = 4.2 × 103) was similar to the calculated value (5.2 × 103), and the molecular weight distribution (Mw/Mn) was only 1.3. In runs 2 and 3, the feed monomer ratio was increased ([M]0/[1]0/[AIBN]0 = 300/8/1 and 700/8/1) and the molecular weights increased to Mn = 11.0 × 103 and 24.0 × 103, respectively. When the monomer was changed to acrylamide (run 4) and N-isopropylacrylamide (run 5), the expected α-alkynyl PAAm and PNIPAM were prepared, respectively.
Table 1 RAFT radical polymerization of vinyl monomers using trithiocarbamate 1
| Run |
Vinyl monomers |
Monomer/CTA/AIBN (molar ratio) |
Temperature (°C) |
Time (h) |
Mn(calculated) (× 10−3) |
Mna (× 10−3) |
Mw/Mna |
Conv.b (%) |
| SEC measurements in CHCl3 relative to poly(styrene) standards; before reprecipitation. Percent conversion determined by 1H NMR spectroscopy. |
| 1 |
Styrene |
100/8/1 |
90 |
20 |
5.2 |
4.2 |
1.3 |
75 |
| 2 |
Styrene |
300/8/1 |
90 |
40 |
15.6 |
11.0 |
1.3 |
54 |
| 3 |
Styrene |
700/8/1 |
90 |
48 |
36.5 |
24.0 |
1.4 |
72 |
| 4 |
Acrylamide |
100/2/1 |
70 |
48 |
3.6 |
3.6 |
— |
83 |
| 5 |
N-Isopropylacrylamide |
100/2/1 |
70 |
24 |
5.7 |
4.8 |
1.7 |
76 |
The MALDI-TOF mass spectrum of the poly(styrene) produced by RAFT radical polymerization using the trithiocarbamate 1 as the RAFT agent was used to determine the absolute molecular weights and the structure of the poly(styrene) (Fig. 3). In the spectrum, there is one set of peaks with a repeat of 104.3 m/z, which corresponds to the molecular weight of the styrene unit. When the peak at 1664 was examined, it was found to coincide with the calculated peak for 1665 [styrene (104 Da) × 12 + α-(R-) and ω-[Z-(CS)S] terminal groups (417 Da) = 1665]. This suggests that the RAFT radical polymerizations were initiated by a fragmented R group from the trithiocarbamate 1; the pattern for a product initiated by AIBN was not observed.
 |
| | Fig. 3 Expanded MALDI-TOF mass spectrum of alkyne-containing PSt. | |
Grafting polystyrene onto aECM-CS5-ELF
To demonstrate a grafting onto aECM protein, aECM-CS5-ELF-N3 (67% incorporation) and α-alkynyl PSt (alkene at α terminus) were coupled, with Cu(I)Br as the catalyst (Scheme 2). The click18 reaction proceeds in DMSO at room temperature for 3 days, affording the expected product in high yield (90–99%). The results are summarized in Table 2. After the PSt was grafted onto the aECM-CS5-ELF-N3, the solubility was altered, and the protein-based graft copolymer was soluble in DMF, DMSO, and even in chloroform (aECM-CS5-ELF-F and aECM-CS5-ELF-N3 are not soluble in chloroform). In the 1H NMR spectrum of (aECM-CS5-ELF-N3)-graft-PSt in CDCl3 (Fig. 4), the signals ascribed to the aECM-CS5-ELF-N3 backbone are clearly visible, although these signals were not detected in the spectrum of aECM-CS5-ELF-N3 (data not shown). The modified solubility in CDCl3 also supports the expected grafting onto the aECM-CS5-N3 protein.
 |
| | Scheme 2 Grafting of alkyne-containing vinyl polymers prepared via RAFT radical polymerization onto extracellular matrix protein containing azide functionality (aECM-CS5-ELF-N3). | |
Table 2 Grafting of vinyl polymers onto aECM-CS5-ELF-N3a
| Run |
Vinyl polymer (Mnb × 10−3) |
[Vinyl polymer]0/[p-N3Phe]0 (molar ratio) |
Solvent |
Temperature (°C) |
Time (day) |
Yieldc (%) |
Conv.d (%) |
| aECM-CS5-ELF-N3 (67% incorporation), in PBS for 4 days. SEC measurements in CHCl3 relative to poly(styrene) standards; before reprecipitation. Calculated based on aECM-CS5-ELF-N3 backbone was quantitative (90–99%). Percent conversion estimated by FT-IR spectroscopy. |
| 1 |
PSt (11.0) |
20/1 |
DMSO |
27 |
4 |
99 |
>99 |
| 2 |
PAAm (3.2) |
10/1 |
PBS |
27 |
4 |
99 |
>99 |
| 3 |
PNIPAM (4.8) |
15/1 |
PBS |
27 |
4 |
99 |
>99 |
 |
| | Fig. 4 1H NMR spectrum of (aECM-CS5-ELF-N3)-graft-PSt (red line) and alkyne-containing PSt (green line) (200 MHz, CDCl3 27 °C). | |
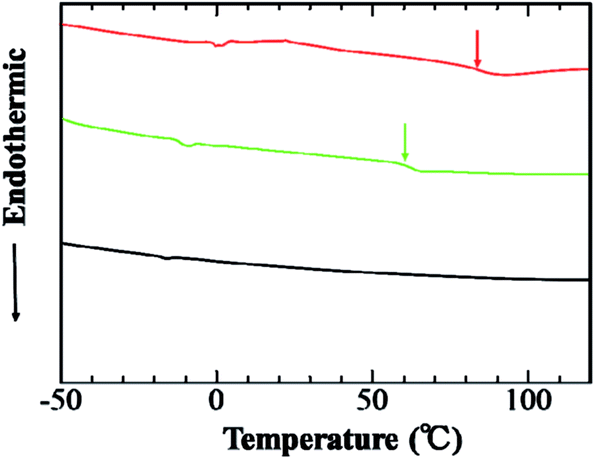
After grafting of PSt onto aECM-CS5-N3, the thermal properties were also changed (Fig. 5). In DSC measurements of the protein-based graft copolymer, the heat capacity has an inflection point ascribed to the Tg at 84 °C. On the other hand, the alkyne-containing PSt (green line) has a Tg at 62 °C. These results also indicate that the expected (aECM-CS5-ELF-N3)-graft-poly(styrene) was synthesized.
 |
| | Fig. 5 DSC trace of (aECM-CS5-ELF-N3)-graft-PSt (red line), alkyne-containing PSt (green line), and aECM-CS5-ELF-N3 (black line) (heating rate: 10 °C min). | |
Grafting of poly(acrylamide) onto aECM-CS5-ELF
In order to demonstrate grafting of hydrophilic vinyl polymer, aECM-CS5-ELF-N3 (67% incorporation) was also coupled with α-alkynyl PAAm (alkyne at α terminus) using Cu(I)Br and THPTA as the catalyst and ligand, respectively (Table 2). The click reaction proceeds in PBS at room temperature for 4 days, also affording the expected product in excellent coupling efficiencies (99%). The grafting was confirmed by disappearance of IR absorbance at 2118 cm−1 ascribed to azido group. Subsequently, the aminolysis was performed using n-propylamine to remove the long alkyl chain (C12H25SCS–), in which UV absorbance due to C12H25SCS– (λ = 305 nm) disappeared (Fig. S2†). In order to cross-check the coupling, both the purified (aECM-CS5-ELF-N3)-graft-PAAm and aECM-CS5-ELF-N3 proteins were analyzed via SDS-PAGE (Fig. 6). The gels show a significant amount of aECM-CS5-ELF-N3 protein with a calculated molecular weight of 42.6 kDa. On the other hand, in the SDS-PAGE of purified (aECM-CS5-ELF-N3)-graft-PAAm, no bands ascribed to proteins (including the parent protein) were observed, indicating that (aECM-CS5-ELF-N3)-graft-PAAm does not show electrophoresis. We speculated that it was due to interaction of PAAm grafts and electrophoretic gel matrix for SDS-PAGE (made of PAAm). From these results, it was concluded that (aECM-CS5-ELF-N3)-graft-poly(acrylamide) was successfully prepared.
 |
| | Fig. 6 SDS-PAGE of aECM-CS5-ELF-N3 (middle) and (aECM-CS5-ELF-N3)-graft-PAAm (right). Left lane is marker. | |
The goal was to produce a protein with an acceptable LCST in order to produce aECM with reversible adhesion. A property of elastin-like polypeptides which distinguishes them from other polypeptides is that they are miscible with water at all concentrations below the lower critical solution temperature (LCST). Furthermore, this miscibility behavior is reversible.11 For aECM-CS5-ELF-N3 (2 mg mL−1; Fig. 7, green line), the LCST value is 36 °C. After grafting of PAAm onto aECM-CS5-ELF-N3, no LCST behavior was observed (Fig. 7, blue line). The secondary structures of elastin-like polypeptides are characterized by a high proportion of random coils and a low proportion of β-turns at low temperatures. The fraction of the secondary structure was calculated by a curve-fitting method using a linear combination of typical CD spectra for α-helical, β-sheet and random coil conformations.20 This distribution of random coils and β-turns gradually inverts with increasing temperature.11 These structural features are reflected in the CD spectra of aECM-CF5-ELF-F at low temperatures by a distinct minimum at 197 nm and a less pronounced minimum at 218 nm, which are associated with the presence of random coils (75%) and β-turns (25%), respectively.11 As expected, with increasing temperature to 60 °C, the random coil signal becomes less pronounced (50%), while that of the β-turn increases (50%), indicating a transition from a less ordered to a more ordered secondary structure state.11
 |
| | Fig. 7 LCST measurements on aECM-CS5-ELF-N3 (green, 67% incorporation), (aECM-CS5-ELF-N3)-graft-PAAm (after aminolysis, blue), and (aECM-CS5-ELF-N3)-graft-PNIPAM (after aminolysis, red LCST = 23 °C) at concentrations of 1 mg mL−1 in H2O. | |
The CD spectra of the two aECMs, aECM-CS5-ELF-N3 (Fig. 8a) and (aECM-CS5-ELF-N3)-graft-PAAm (Fig. 8b) demonstrate that they retain the characteristics associated with proteins having mixed random coil (43 and 47%, respectively)/β-turns (16 and 10%, respectively), although in both cases there appears to be an increased amount of α-helices (41 and 43%, respectively) (as demonstrated by the presence of signals at 206 nm and 222 nm) at low temperature (20 °C) in comparison with aECM-CS5-ELF-F.11 In aECM-CS5-ELF-N3, the random coil signal (43% at 20 °C) is remarkably decreased (17%) as the temperature increases to 60 °C (Fig. 8a), and thus, the CD spectra no longer resemble proteins having random coil/β-turn secondary structures.11 Instead, the proportion of β-turns increased to 47%, inducing LCST behavior (Fig. 8a). After grafting (Fig. 8b), the spectrum was similar to that of aECM-CS5-ELF-N3 and showed double minima at 204 and 222 nm, indicating an increased proportion of α-helices (43%) at 20 °C. However, the spectra were unaffected by temperature (Fig. 8b), in that a high proportion of random coils (42%) and α-helices (42%) and a low proportion of β-turns (16%) persist even at 60 °C. These results indicate that the introduction of PAAm side chains does not alter the secondary structure of the parent protein (aECM-CS5-ELF-N3) and the disappearance of the LCST response is ascribed not to the conformation of the backbone but the increased hydrophilicity ascribed to the PAAm side chains.
 |
| | Fig. 8 CD spectra of aECM-CS5-ELF-N3 (a), and (aECM-CS5-ELF-N3)-graft-PAAm (b) at a concentration of 0.01 mg mL−1 (a) and 0.05 mg mL−1 (b) in H2O. | |
Grafting poly(N-isopropylacrylamide) onto aECM-CS5-ELF
The ultimate goal is the temperature-dependent detachment of cultivated and spread HUVECs, because the (aECM-CS5-ELF-N3)-graft-PNIPAM has LCST as well as a cell binding site. Okano et al. already reported the temperature-dependent detachment of cultivated and spread cells using a PNIPAM surface, in which the hydration/dehydration of the PNIPAM segment switched the detachment/adhesion.15 Following the similar procedure described above, grafting onto aECM-CS5-ELF-N3 was performed to afford the expected (aECM-CS5-ELF-N3)-graft-PNIPAM. Similar to (aECM-CS5-ELF-N3)-graft-PAAm, the grafting was confirmed by disappearance of IR absorbance at 2118 cm−1 ascribed to azido group and successively the aminolysis was performed using n-propylamine to remove the long alkyl chain (C12H25SCS–), in which UV absorbance due to C12H25SCS– (λ = 305 nm) disappeared (Fig. S2†). After grafting, (aECM-CS5-ELF-N3)-graft-PNIPAM showed LCST behavior at 22 °C ascribed to PNIPAM segments (Fig. 7, please see also Fig. S3†), while (aECM-CS5-ELF-N3)-graft-PAAM did not (Fig. 7). The structure of (aECM-CS5-ELF-N3)-graft-PNIPAM was also confirmed by 1H NMR in DMSO-d6 (Fig. S4†).
Photo cross-linking of (aECM-CS5-ELF-N3)-graft-PAAm
As (aECM-CS5-ELF-N3)-graft-PAAm has no remaining azide functionality because the click grafting was completed, the coated film (a physical mixture of aECM-CS5-ELF-N3 and (aECM-CS5-ELF-N3)-graft-PAAm) was irradiated (λ = 250–400 nm) to demonstrate photo cross-linking to afford the coated blend film. After UV irradiation, the coated film was not soluble in organic or aqueous media. Photo cross-linking of a physical mixture of aECM-CS5-ELF-N3 and (aECM-CS5-ELF-N3)-graft-PNIPAM was also performed and similar coated film was obtained.
Cell spreading
According to previous papers,6b,6c,11 cell spreading on cross-linked aECM film is dependent on the presence of REDV domains. HUVECs plated on cross-linked aECM-CS5-ELF-N3/(aECM-CS5-ELF-N3)-graft-PAAm blend films were monitored at 1 day intervals by phase contrast microscopy and were found to be dark (spread) (Fig. 9). The number of adhered cells was actually counted using UV measurements. After microscopic observation, the cells were stained with 0.5% aqueous crystal violet11,21 for 10 min in order to quantify the adhered cells. Following the reported procedure,11,21 the dishes were washed with water to remove the bright, rounded (not spread) cells, which were then dried at 37 °C. Subsequently, 1% sodium dodecyl sulfate was added, the cells were dissolved for 1 h, and the absorbance at 595 nm (ascribed to crystal violet from spread cells) was estimated using UV/vis spectroscopy (Fig. 10). On blended films, in which the p-N3Phe unit acts as the photo-sensitive cross linker via nitrene chemistry, the cells spread within 1 day (Fig. 9). When compared with a positive control cultured on a commercially available cultivation dish [Cell Desk (Nunc, Inc.), Fig. 9b], the percentage of well spread cells on cross-linked (aECM-CS5-ELF-N3)-graft-PAAm (>99% grafting) (Fig. 9d) was significantly higher at all times, indicating that the REDV segment acts as a cell-binding segment. On the other hand, (aECM-CS5-ELF-N3)-graft-PNIPAM, prepared via click reaction, also showed adhesion of HUVECs (Fig. 9e), but introduction of the PNIPAM content resulted in a significant reduction in the extent of cell spreading (Fig. 9e). We speculated that extent of cell spreading upon grafting of PAAm was higher than that of PNIPAM, because at 37 °C the former is more swollen with cultivation media than latter.
 |
| | Fig. 9 Cell morphology after 10 min to 4 days on (a) a blank, (b) a positive control [commercially available cultivation dish], (c) cross-linked aECM-CS5-ELF-N3, (d) a cross-linked blend of aECM-CS5ELF-N3/(aECM-CS5-ELF-N3)-graft-PAAm, and (e) a cross-linked blend of aECM-CS5ELF-N3/(aECM-CS5-ELF-N3)-graft-PNIPAM. | |
 |
| | Fig. 10 Competitive peptide inhibition of HUVEC adhesion onto aECMs with 1.5 mM GREDVY. | |
Competitive peptide inhibition
To test the hypothesis that the HUVECs adhere to the cross-linked film through the REDV cell-binding domain and that non-specific cell adhesion can be suppressed by grafting of PAAm or PNIPAM, competitive peptides were used to inhibit adhesion. Tirrell et al. previously showed that cell adhesion to absorbed aECM proteins is dependent upon the presentation of authentic cell-binding domains.6 However, cross-linked protein films exhibit significant nonspecific adhesion.6 After the HUVECs were incubated on the cross-linked film, subsequent washing with the competitive GREDVY21 peptide (1.5 mM in PBS) was carried out. GREDVY21 peptide was prepared in our laboratory and the structure was confirmed by MALDI-TOF mass spectrum (H2N-GREDVY-COOH + Na+: m/z = 762.2) (Fig. S5†). The number of cells adhered to cross-linked aECM-CS5-ELF-N3 (HUVECs were incubated on the cell culture Petri dish) was counted and shown in Fig. 10, in which in the absence of peptide to normalize (expressed as 100%) for passage-to-passage variations. After washing, significant non-specific adhesion was observed in the cross-linked aECM-CS5-ELF-N3 (86% on average) and (aECM-CS5-ELF-N3)-graft-PAAm (90% on average) films, but non-specific adhesion was apparently decreased (12% on average) in the cross-linked (aECM-CS5-ELF-N3)-graft-PNIPAM film (Fig. 10). The negative control peptide GRDEVY (not GREDVY) had no significant effect on the number of cells adhered to aECM. Furthermore, increasing the GREDVY concentration from 1.5 mM to 3.0 mM decreased the number of HUVECs adhered to the film. These results demonstrate that some HUVECs specifically adhere to the REDV cell-binding domain in the cross-linked film, and that non-specific adhesion is suppressed by the introduction of PNIPAM side chains via a “grafting onto” procedure.
Cell detachment with temperature control
For the cross-linked blend of aECM-CS5ELF-N3/(aECM-CS5-ELF-N3)-graft-PNIPAM, cell detachment was also evaluated by decreasing the temperature below the LCST (15 °C) after incubation at 37 °C for 4 days. The HUVECs were observed at 15 °C for 60 min (Fig. 11). The results indicate that the surfaces prepared from cross-linked aECM-CS5-ELF-N3 and a cross-linked blend of aECM-CS5ELF-N3/(aECM-CS5-ELF-N3)-graft-PNIPAM were effective at triggering cell detachment when the temperature fell below the LCST (Fig. 11). On the aECM-CS5ELF-N3/(aECM-CS5-ELF-N3)-graft-PAAm surface, few cells became round after the observation period (60 min, Fig. 11c), which coincides well with the lack of thermosensitivity (the LCST behavior shown in Fig. 7). Fig. 11b shows a set of phase contrast images of an area of HUVECs in real-time from 0 to 60 min at ca. 15 °C. At first, most cells adopted a spindle shape with the formation of lamellipodia. After 60 min, most cells adopted a round morphology as they had lost their anchoring points. It should be noted that some HUVECs failed to detach from the surface because of the absence of intracellular interactions (as observed in cell sheets), but they could be detached by gently rinsing with the culture medium. Similar to described above, 1% sodium dodecyl sulfate was added, the cells were dissolved for 1 h, and the absorbance at 595 nm (ascribed to crystal violet from spread cells) was estimated using UV/vis spectroscopy (Fig. 12). Thus, the rounded cells observed were also counted as detached cells in this work (Fig. 12). Fig. 12 indicated the HUVEC cells adhered counted by UV measurement and that the cross-linked blend of aECM-CS5ELF-N3/(aECM-CS5-ELF-N3)-graft-PNIPAM was the most sensitive for temperature-triggered cell detachment in this experimental condition.
 |
| | Fig. 11 Time lapse microscopic imaging of HUVECs detaching from (a) cross-linked aECM-CS5-ELF-N3, (b) a cross-linked blend of aECM-CS5-ELF-N3/(aECM-CS5-ELF-N3)-graft-PNIPAM, and (c) a cross-linked blend of aECM-CS5-ELF-N3/(aECM-CS5-ELF-N3)-graft-PAAm prepared by dip coating. A progressive cell shrinking and detaching process is observed over 1 h. | |
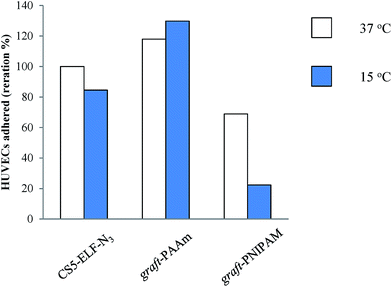 |
| | Fig. 12 HUVEC detachment from (left) cross-linked aECM-CS5-ELF-N3, (middle) a cross-linked blend of aECM-CS5ELF-N3/(aECM-CS5-ELF-N3)-graft-PNIPAM, and (right) a cross-linked blend of aECM-CS5ELF-N3/(aECM-CS5-ELF-N3)-graft-PAAm prepared by dip coating. | |
Conclusions
The results presented herein provide a new tool for the de novo design of “intelligent aECMs” that may be used in cell cultivation scaffolds as well as small-diameter vascular graft construction. Vinyl polymers chemically coupled to Phe analogs incorporated into aECM-CS5-ELF-N3 via click reactions can fine-tune the adhesion behavior of HUVEC/aECM-CS5-ELF systems. These fundamental results provide new guidelines for the design of cultivation scaffolds based on hydration/dehydration (LCST) behavior.
Acknowledgements
A.T. is grateful to Prof. David A. Tirrell for the opportunity to work in his laboratory at California Institute of Technology. The authors acknowledge support from the Nagoya Institute of Technology's Research Promotion Program, from NIH grants EB1971 and GM 62523, and from the NSF Center for the Science and Engineering of Materials at the California Institute of Technology. We thank Dr Hideo Yoshizato for technical assistance.
References
- F. G. Giancotti and E. Ruoslahti, Science, 1999, 285, 1028–1032 CrossRef CAS.
- S. M. Schoenwaelder and K. Burridge, Curr. Opin. Cell Biol., 1999, 11, 274–286 CrossRef CAS.
- M. J. Humphries and P. Newham, Trends Cell Biol., 1998, 8, 78–83 CrossRef CAS.
- R. O. Hynes, Fibronectins, Springer, New York, 1990 Search PubMed.
- Fibronectins, ed. D. F. Mosher, Academic Press, New York, 1989 Search PubMed.
-
(a) A. Panitch, T. Yamaoka, M. J. Fournier, T. L. Mason and D. A. Tirrell, Macromolecules, 1999, 32, 1701 CrossRef CAS;
(b) E. R. Welsh and D. A. Tirrell, Biomacromolecules, 2000, 1, 23 CrossRef CAS;
(c) S. C. Heilshorn, K. A. DiZio, E. R. Welsh and D. A. Tirrell, Biomaterials, 2003, 24, 4245 CrossRef CAS;
(d) P. J. Nowatzki and D. A. Tirrell, Biomaterials, 2004, 25, 1261–1267 CrossRef CAS;
(e) J. C. Liu, S. C. Heilshorn and D. A. Tirrell, Biomacromolecules, 2004, 5, 497–504 CrossRef CAS PubMed;
(f) J. C. Liu and D. A. Tirrell, Biomacromolecules, 2008, 9, 2984–2988 CrossRef CAS PubMed.
- E. Dejana, S. Colella, G. Conforti, M. Abbadini, M. Gaboii and P. C. Marchisio, J. Cell Biol., 1988, 107, 1215–1223 CrossRef CAS.
-
(a) M. J. Humphries, S. K. Akiyama, A. Komoriya, K. Olden and K. M. Yamada, J. Cell Biol., 1986, 103, 2637–2647 CrossRef CAS;
(b) S. K. Akiyama, A. Komoriya, K. M. Yamada and M. J. Humphries, J. Biol. Chem., 1991, 266, 3579–3585 Search PubMed.
-
(a) J. C. M. van Hest and D. A. Tirrell, Chem. Commun., 2001, 1897–1904 RSC;
(b) K. L. Kiick, C. M. van Hest and D. A. Tirrell, Angew. Chem., Int. Ed., 2000, 39, 2148 CrossRef CAS;
(c) P. Wang, Y. Tang and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 6900 CrossRef CAS PubMed;
(d) I. Kwon, K. Kirshenbaum and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 7512 CrossRef CAS PubMed;
(e) M. Mock, T. Michon and D. A. Tirrell, Polym. Prepr., 2003, 44(1), 1065 CAS.
-
(a) K. Kirshenbaum, I. S. Carrico and D. A. Tirrell, ChemBioChem, 2002, 3, 235–237 CrossRef;
(b) D. Datta, P. Wang, I. S. Carrico, S. L. Mayo and D. A. Tirrell, J. Am. Chem. Soc., 2002, 124, 5652 CrossRef CAS PubMed;
(c) I. S. Carrico, S. A. Maskarinec, S. C. Heilshorn, M. L. Mock, J. C. Liu, P. J. Nowatzki, C. Franck, G. Ravichandran and D. A. Tirrell, J. Am. Chem. Soc., 2007, 129, 4874–4875 CrossRef CAS PubMed.
- A. Takasu, S. Kondo, A. Ito, Y. Furukawa, M. Higuchi, T. Kinoshita and I. Kwon, Biomacromolecules, 2011, 12, 3444–3452 CrossRef CAS PubMed.
- F. W. Studier and B. A. Moffatt, J. Mol. Biol., 1986, 189, 113–130 CrossRef CAS.
- A. H. Rosenberg, B. N. Lade, D. Chui, S. Lin, J. J. Dunn and F. W. Staudier, Gene, 1987, 56, 125–135 CrossRef CAS.
- F. W. Studier, A. H. Rosenberg, J. J. Dunn and J. W. Dubendorff, Methods Enzymol., 1990, 185, 60–89 CAS.
- N. Yamada, T. Okano, H. Sakai, F. Karikusa, Y. Sawasaki and Y. Sakurai, Makromol. Chem., Rapid Commun., 1990, 11, 571 CrossRef CAS PubMed.
-
(a) D. Mortisen, M. Peroglio, M. Alini and D. Eglin, Biomacromolecules, 2010, 11, 1261–1272 CrossRef CAS PubMed;
(b) C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS PubMed;
(c) P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS PubMed.
-
(a) B. S. Sumerlin, ACS Macro Lett., 2012, 1, 141–145 CrossRef CAS;
(b) N. Vanparijs, S. Maji, B. Louage, L. Voorhaar, D. Laplace, Q. Zhang, Y. Shi, W. E. Hennink, R. Hoogenboom and B. G. De Geest, Polym. Chem., 2015 10.1039/c4py01224k.
-
(a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS;
(b) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed;
(c) S. Hvilsted, Polym. Int., 2012, 61, 485–494 CrossRef CAS PubMed.
- R. Ranjan and W. J. Brittain, Macromolecules, 2007, 40, 6217–6223 CrossRef CAS.
- N. Greenfield and G. D. Fasman, Biochemistry, 1969, 8, 4108–4116 CrossRef CAS.
- F. Yokoyama, N. Suzuki, M. Haruki, N. Nishi, S. Oishi, N. Fujii, A. Utani, H. K. Kleinman and M. Nomizu, Biochemistry, 2004, 43, 13590–13597 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, UV, MALDI-TOF mass, and IR spectra. See DOI: 10.1039/c5ra04643b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.