
A family of unsymmetrical hydroxyl-substituted BEDT-TTF donors: syntheses, structures and preliminary thin film studies†
Qiang Wangab,
Matteo Zecchinic,
John D. Wallisc,
Yiliang Wude,
Jeremy M. Rawsonf and
Melanie Pilkington*a
aDepartment of Chemistry, Brock University, 500 Glenridge Avenue, Ontario L2S 3A1, Canada. E-mail: mpilkington@ubrock.ca
bSchool of Chemistry and Chemical Engineering, Wuhan Textile University, Wuhan, 430073, China
cSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, UK
dXerox Research Centre of Canada, 2660 Speakman Drive, Mississauga, ON L5K 2L1, Canada
eCorporate Technology, TE Connectivity, 306 Constitution Drive, Menlo Park, CA 94025, USA
fDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Ave., Windsor, Ontario N9B 3P4, Canada
First published on 14th April 2015
Abstract
Three new unsymmetrical hydroxyl-functionalized donors H1–H3 closely related to hydroxymethyl-BEDT-TTF have been synthesised and characterised. Cyclic voltammetry studies showed that the compounds exhibit reversible two one-electron redox processes typical for BEDT-TTF derivatives. X-ray diffraction studies of H1 and H2 reveal π-stacking interactions between pairs of donors that are organized into distinct H-bonded square motifs and DFT calculations indicate that the HOMO is located on the central 1,3-dithiole rings. Protection of the hydroxyl group with acetyl in 13 eliminates co-facial S⋯S interactions between the dimers to accommodate the bulkier side chains, but short edge-to-edge S⋯S contacts offer an alternative pathway for electron mobility. Chemical oxidation of H1 and HMET 2 with I2 afforded single crystals of two 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 charge transfer salts, 18 and 19. The molecules pack as dimers with close π-stacking interactions between pairs of radical cations whose crystal structures are further stabilized via an interplay of S⋯S and S⋯I contacts. Iodine-doped surface conducting polystyrene blend films of H3 deposited on a silica substrate exhibit quasiconducting properties, but afford no OFET response when fabricated into devices. Visible-NIR studies of a doped polystyrene blend film of H3 cast on a glass substrate show absorption bands at λ = 950 and 3000 nm, consistent with mixed valence states due to the presence of charge-transfer species on the surface of the films.
:1 charge transfer salts, 18 and 19. The molecules pack as dimers with close π-stacking interactions between pairs of radical cations whose crystal structures are further stabilized via an interplay of S⋯S and S⋯I contacts. Iodine-doped surface conducting polystyrene blend films of H3 deposited on a silica substrate exhibit quasiconducting properties, but afford no OFET response when fabricated into devices. Visible-NIR studies of a doped polystyrene blend film of H3 cast on a glass substrate show absorption bands at λ = 950 and 3000 nm, consistent with mixed valence states due to the presence of charge-transfer species on the surface of the films.
Introduction
First used to prepare an organic metal in the 1970's,1 tetrathiafulvalene (TTF) and its derivatives now represent one of the most well studied classes of sulfur containing heterocycles of interest to main group, organic, supramolecular and materials chemists worldwide.2 In more recent years this family of compounds have found applications as molecular switches3 shuttles and sensors,4 as well as redox accessible organic donors for the preparation of conducting radical cation salts.5 The driving force in the crystallization of the salts are their strong π–π stacking interactions that, together with C–H⋯S and S⋯S contacts, facilitate the intermolecular electronic transfer responsible for their transport properties. Until fairly recently conducting salts of TTFs were prepared as single crystals with mobilities exceeding 10 cm2 V−1 s−1 for modified derivatives with side chains. Although crystalline radical cation salts have found applications in the field of organic electronics,6 attention has been more recently focused on developing more soluble derivatives for solution-processable technologies.7 One clear advantage of this approach is that it affords tuneable materials that combine the unique electronic properties of molecular metals (e.g. metallic conductivity) together with the favourable properties of a polymeric matrix for applications where low-cost, large-area coverage and flexibility are important considerations.In contrast to TTF, the chemistry of bis(ethylenedithio)-tetrathiafulvalene, more commonly known as BEDT-TTF or ET, has been much less well explored even though it has played a prominent role in the development of molecular conductors, superconductors, and bifunctional materials.8 Although formation of conducting films of BEDT-TTF have been studied,9 reports of films prepared from its substituted derivatives are restricted to the formation of LB films from hexadecyl-(BEDT-TTF), and its combination with hexadecyl-TCNQ, or reaction with Fe(III).10 Related studies have also involved deposition of a mercaptodecylthio-(EDT-TTF) derivative onto a mica supported gold surface,11 and hydroxymethyl substituted EDT-TTF derivatives have been combined with arachidonic acid to form weakly conducting Langmuir–Blodgett films.12
Over the last two decades our research program has predominantly focused on developing synthetic methodologies for the preparation of new families of BEDT-TTF derivatives, particularly chiral donors.13 In more recent years we have shifted our attention to establish libraries of organic donors whose electronic properties and solubilities can be tuned via molecular design to render them suitable for a particular electronic application. In this context we recently reported the electronic properties of thin films prepared from thiophene appended BEDT-TTF derivatives e.g. 1 for applications as organic field effect transistors (OFETs).14 In this new study we report the synthesis and study of a new family of unsymmetrical hydroxyl-substituted donors H1–H3, the O-acetyl protected intermediate 13, and the previously reported hydroxymethyl BEDT-TTF 213d (Fig. 1). Hydrogen bonds are one of the fundamentally important non-covalent interactions in chemistry and biology that have also recently been shown to play a role in the switching of conductivity and magnetism in a TTF derivative.15 Our objectives were two fold; firstly to introduce hydroxyl substituents into the molecular framework of the BEDT-TTF donor to optimize the number and type of intermolecular interactions, increase the dimensionality of the materials and thus improve their charge transport properties; secondly, we set out to tune the solubility of the donors and establish experimental conditions for the preparation of solution processable thin films in order to evaluate their electrical properties and suitability for OFET applications.
 | ||
| Fig. 1 Molecular structures of TTF, BEDT-TTF, the thiophene appended donor 1, HMET 2 and the hydroxyl donors H1–H3. | ||
Experimental
General considerations
All experiments were performed under a nitrogen atmosphere unless stated otherwise. Dry solvents were obtained from a Puresolve PS MD-4 solvent purification system. 1H and 13C NMR spectra were recorded on Bruker AVANCE AV300 or AV600 NMR spectrometers and chemical shifts were determined with reference to residual solvent. IR spectra were recorded on a Mattson Research Series FT-IR spectrometer as KBr discs. EI and HR FAB mass spectrometry measurements were obtained from a KRATOS/MSI CONCEPT 1-S spectrometer. Accurate mass, nanoelectrospray measurements were obtained on an LTQ Oribtrap XL spectrometer. Elemental analyses were obtained from Atlantic Microlab. Melting points were measured on a SMP10 melting point apparatus. Cyclic voltammetry measurements were recorded at room temperature under N2 in a conventional three-electrode cell using Pt working electrodes (3 mm diameter), a Pt wire counter electrode, an Ag/AgCl reference electrode and a BAS Epsilon potentiostat. Electronic absorption spectra were measured on a Varian 5000 UV-vis-NIR spectrophotometer. Fabricated OTFT devices were evaluated using a Keithley SCS-4200 characterization system under ambient conditions. Four-probe DC measurements were carried out on a Keithley 236 Source Measurement Unit. HMET was prepared according to literature procedures.13d Experimental details for the synthesis of the O-acetyl protected compounds 7, 13 and 17 together with donors H1 to H3 are presented in the text. Full experimental procedures for the preparation of all synthetic intermediates are provided in S-1 of the ESI.†Synthesis of (2-acetoxypropylene-1,3-dithio)(ethylenedithio) tetrathiafulvalene (7)
A suspension of oxo compound 5 (0.70 g, 2.50 mmol) and unsubstituted thione 6 (1.12 g, 5.00 mmol) in dry triethyl phosphite (5 mL) was heated to 90 °C under nitrogen for 16 h. The mixture was cooled to room temperature and hexane (10 mL) was added to facilitate further precipitation. The solid was collected and washed with hexane (5 mL). The crude product was purified by flash chromatography on silica (hexane:CH2Cl2 1:2) to give first BEDT-TTF and then compound 7 as an orange-yellow solid (0.75 g, 66%), m.p. 185–187 °C. δH (300 MHz, CDCl3): 5.24 (1H, m, 2-H), 3.31 (4H, s, 5′-,6′-H2), 2.96 (2H, dd, J = 13.7, 2.1 Hz, 1-,3-Hα), 2.65 (2H, dd, J = 13.7, 9.9 Hz, 1-,3-Hβ), 2.11 (3H, s, CH3); δC (75 MHz, CDCl3): 169.48 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 130.32, 113.79, 113.29 & 111.66 (6 × sp2C), 77.22 (2-C), 36.04 (1-,3-C), 30.16 (–CH2CH2–), 21.06 (CH3); IR νmax (cm−1, KBr): 2964 (w), 2920 (w), 1740 (s), 1417 (w), 1292 (w), 1230 (vs), 1109 (w), 1014 (m), 957 (w), 889 (w), 771 (w), 648 (w), 505 (w); m/z: (EI+) 456 ([M]+, 50%); HRMS: (EI) found [M]+ 455.85995, C13H12O2S8 requires 455.86031. Further elution afforded the homo coupled cis and trans isomers of bis(2-acetoxypropylene-1,3-dithio) tetrathiafulvalene 8 as an orange solid (70 mg, 5%), m.p. 270–273 °C (dec); δH: (300 MHz, CDCl3): 5.22 (m, 2H, 2-,2′-H), 2.94 (4H, dd, J = 14.4, 2.7 Hz, 1-,1′-,3-,3′-Hα), 2.66 (4H, m, 1-,1′-,3-,3′-Hβ), 2.11 & 2.10 (6H, 2 × CH3); δC: (75 MHz, CDCl3): 169.60 & 169.49 (CO, 2 isomers), 130.30 & 112.42 (6 × sp2C), 79.91 (2-,2′-C), 36.12 & 36.00 (1-,1′-,3-,3′-C, 2 isomers), 21.11 (2 × CH3); IR νmax (cm−1, KBr): 2968 (w), 2920 (w), 1740 (vs), 1423 (w), 1403 (w), 1369 (m), 1232 (vs), 1111 (w), 1020 (s), 960 (m), 903 (m), 770 (w), 646 (w), 501 (w); m/z: (FAB+) 528 ([M]+, 100%); HRMS: (FAB) found [M]+ 527.88348, C16H16O4S8 requires 527.88144.
O), 130.32, 113.79, 113.29 & 111.66 (6 × sp2C), 77.22 (2-C), 36.04 (1-,3-C), 30.16 (–CH2CH2–), 21.06 (CH3); IR νmax (cm−1, KBr): 2964 (w), 2920 (w), 1740 (s), 1417 (w), 1292 (w), 1230 (vs), 1109 (w), 1014 (m), 957 (w), 889 (w), 771 (w), 648 (w), 505 (w); m/z: (EI+) 456 ([M]+, 50%); HRMS: (EI) found [M]+ 455.85995, C13H12O2S8 requires 455.86031. Further elution afforded the homo coupled cis and trans isomers of bis(2-acetoxypropylene-1,3-dithio) tetrathiafulvalene 8 as an orange solid (70 mg, 5%), m.p. 270–273 °C (dec); δH: (300 MHz, CDCl3): 5.22 (m, 2H, 2-,2′-H), 2.94 (4H, dd, J = 14.4, 2.7 Hz, 1-,1′-,3-,3′-Hα), 2.66 (4H, m, 1-,1′-,3-,3′-Hβ), 2.11 & 2.10 (6H, 2 × CH3); δC: (75 MHz, CDCl3): 169.60 & 169.49 (CO, 2 isomers), 130.30 & 112.42 (6 × sp2C), 79.91 (2-,2′-C), 36.12 & 36.00 (1-,1′-,3-,3′-C, 2 isomers), 21.11 (2 × CH3); IR νmax (cm−1, KBr): 2968 (w), 2920 (w), 1740 (vs), 1423 (w), 1403 (w), 1369 (m), 1232 (vs), 1111 (w), 1020 (s), 960 (m), 903 (m), 770 (w), 646 (w), 501 (w); m/z: (FAB+) 528 ([M]+, 100%); HRMS: (FAB) found [M]+ 527.88348, C16H16O4S8 requires 527.88144.
Synthesis of (ethylenedithio)(2-hydroxypropylene-1,3 dithio) tetrathiafulvalene (H1)
A solution of protected donor 7 (0.55 g, 1.21 mmol) in THF (30 mL) and 6 M HCl solution (6.5 mL) was stirred under nitrogen for 40 h. The solution was neutralized by the addition of solid NaHCO3. The organic layer was collected, washed with brine and dried over MgSO4. Removal of solvent yielded H1 as an orange-yellow solid (0.48 g, 96%), m.p. 227–230 °C (dec). δH (600 MHz, DMSO-d6): 5.62 (1H, d, J = 3.6 Hz, OH), 3.96 (1H, m, 2-H), 3.41 (4H, s, 5′-,6′-H2), 2.96 (2H, dd, J = 14.4, 2.4 Hz, 1-,3-Hα), 2.46 (2H, dd, J = 13.2, 9.6 Hz, 1-,3-Hβ); δC (150 MHz, DMSO-d6): 129.88, 113.26, 109.18 (6 × sp2-C), 73.50 (2-C), 38.58 (1-,3-C), 29.99 (–CH2CH2–); IR νmax (cm−1, KBr): 3547 (s), 3392 (w), 3313 (w), 2958 (w), 2910 (w), 1728 (w), 1645 (w), 1551 (w), 1412 (m), 1290 (w), 1165 (m), 1057 (w), 1013 (vs), 918 (w), 889 (m), 770 (m), 743 (m), 507 (w), 463 (w). m/z: (EI+) 414 ([M]+, 100%); HRMS: (EI) found [M]+ 413.85016, C11H10OS8 requires 413.84975. Elem. anal. found C: 31.76, H: 2.50%. C11H10OS8 requires C: 31.88, H: 2.43%.Synthesis of (cis-4′′,4′′-bis(acetoxymethyl)cyclopenta-1,2-dithio) (ethylene-dithio)tetrathiafulvalene (13)
A suspension of oxo compound 12 (1.72 g, 4.40 mmol) and unsubstituted thione 6 (1.98 g, 8.80 mmol) in dry triethyl phosphite (6 mL) was heated to 90 °C under nitrogen for 16 h. The mixture was cooled to RT and hexane (40 mL) was added to facilitate further precipitation. The solid was collected by filtration and washed with hexane (5 mL). The residue obtained was purified by a flash chromatography on silica eluting with CH2Cl2 to give 13 as an orange crystalline solid (1.31 g, 53%), m.p. 168–170 °C. δH (300 MHz, CDCl3): 4.06 (2H, s), & 3.97 (2H, s) (2 × 4′′-CH2O), 3.92 (2H, m, 1′′-,2′′-H), 3.33 (4H, s, 5′-,6′-H2), 2.15 (2H, m, 3′′-,5′′-Hα), 2.10 (3H, s) & 2.09 (3H, s) (2 × CH3), 1.86 (2H, m, 3′′-,5′′-Hβ); δC (75 MHz, CDCl3): 171.01 (2 × CO), 124.51, 113.78, 112.56 & 112.13 (6 × sp2C), 67.40 & 65.88 (2 × 4′′-CH2O), 52.31 (1′′-,2′′-C), 46.18 (4′′-C), 38.43 (3′′-,5′′-C), 30.15 (5′-,6′-H2), 20.81 (2 × CH3); IR νmax (cm−1, KBr): 2947 (w), 2924 (w), 2854 (w), 1732 (vs), 1433 (w), 1367 (m), 1236 (vs), 1038 (s), 987 (w), 912 (w), 889 (w), 768 (w), 679 (w), 606 (w), 449 (w); m/z: (FAB) 568 ([M]+, 100%); HRMS: (FAB) found [M]+ 567.91530, C19H20O4S8 requires 567.91274.
Synthesis of (cis-4′′,4′′-bis(hydroxymethyl)cyclopenta-1,2-dithio) (ethylene-dithio)tetrathiafulvalene (H2)
A solution of the bis(acetyl) protected donor 13 (1.17 g, 2.06 mmol) in THF (50 mL) and 6 M HCl solution (22 mL) was stirred under N2 for 43 h. THF (80 mL) was added and the mixture was neutralized by the addition of solid NaHCO3. The organic layer was collected and dried over MgSO4. The crude product was purified by chromatography on silica firstly eluted with THF:hexane (3:2) to remove side products, followed by THF to elute H2. Further product was obtained by thorough extraction of silica from the top of the column with THF, evaporation and washing the solid with CH2Cl2. H2 was obtained as a yellow solid (0.65 g, 65%), m.p. 211–213 °C (dec). δH (600 MHz, DMSO-d6): 4.67 (s, 2H, 2 × OH), 4.01 (2H, m, 2 × 1′′,2′′-H), 3.41 (4H, s, 5′-,6′-H2), 3.28 (2H, s) & 3.25 (2H, s) (2 × 4′′-CH2O), 1.95 (2H, m, 3′′-,5′′-Hα), 1.61 (2H, m, 3′′-,5′′-Hβ); δC (150 MHz): 123.01, 113.22, 111.70 & 111.38 (6 × sp2C), 65.90 & 64.52 (2 × 4′′-CH2O), 52.14 (1′′-,2′′-C), 49.87 (4′′-C), 37.74 (3′′-,5′′-C), 29.95 (5′-,6′-H2); IR νmax (cm−1, KBr): 3305 (vs, br), 2918 (m), 2868 (m), 1645 (w), 1551 (w), 1439 (m), 1406 (m), 1284 (m), 1255 (w), 1198 (m), 1146 (w), 1088 (w), 1041 (vs), 1018 (vs), 908 (m), 771 (m), 679 (w), 577 (w), 473 (w); m/z: (FAB) 484 ([M]+, 60%); m/z: (EI+) 484 ([M]+, 10%); HRMS: (EI) found [M]+ 483.89134, C15H16O2S8 requires 483.89161; elem. anal. found C: 36.38, H: 3.13%; C15H16O2S8·0.25CH2Cl2 requires C: 36.22, H: 3.27%.
(±) (1′′R,5R)- and (1′′R,5S) (1′′-acetoxybutyl)bis-(ethylenedithio)tetrathiafulvalene (17)
A suspension of oxo compound 16 (1.54 g, 4.78 mmol) and unsubstituted thione 6 (2.13 g, 9.56 mmol) in dry triethyl phosphite (7 mL) was heated to 90 °C under nitrogen for 24 h. The mixture was cooled to RT and triethyl phosphite was removed by distillation under reduced pressure. The residue was purified by flash chromatography on silica (hexane:DCM 1:1) to give 17 as an orange solid (1.34 g, 56%). 1H NMR showed the product was a mixture of diastereomers with estimated ratio 65:35. m.p. 78–80 °C; δH (300 MHz, CDCl3): 5.20 (0.35H, q, J = 6.1 Hz, 1′′-H), 5.15 (0.65H, q, J = 6.1 Hz, 1′′-H), 3.79 (1H, m, 5-H), 3.31 (4H, s, 5′-,6′-H2), 3.20 (1H, m, 6-Hα), 3.08 (1H, m, 6-Hβ), 2.11 (3H, s, CH3CO), 1.73 (2H, m, 2′′-H2), 1.37 (2H, m, 3′′-H2), 0.96 (3H, t, J = 7.3 Hz, 4′′-H3); δC (75 MHz): 170.34 & 170.18 (CO), 115.60, 115.22, 114.99 & 113.84 (6 × sp2C), 73.68 & 73.63 (1′′-C), 49.49 & 48.78 (5-C), 33.89 & 33.81 (2′′-C), 32.95 & 32.22 (6-C), 30.20 (5′-,6′-C), 20.94 (CH3CO), 18.57 & 18.36 (3′′-C), 13.84 & 13.79 (4′′-C); IR νmax (cm−1, KBr): 2954 (w), 2922 (w), 2866 (w), 1738 (s), 1458 (w), 1410 (w), 1367 (m), 1227 (vs), 1120 (w), 1020 (m), 887 (w), 770 (m), 635 (w), 606 (w), 490 (w); m/z: (FAB+) 498 ([M]+, 100%); HRMS: (FAB) found [M]+ 497.90754, C16H18O2S8 requires 497.90726.
Synthesis of (±) (1′′R,5R)- and (1′′R,5S)-(1′′-hydroxybutyl) bis(ethylenedithio)tetrathiafulvalene (H3)
A solution of ester 17 (1.30 g, 2.61 mmol) in THF (30 mL) and 6 M HCl solution (16 mL) was stirred under nitrogen for 40 h. The solution was neutralized by the addition of solid NaHCO3. The organic layer was collected, washed with brine and dried over MgSO4. Removal of solvent yielded a sticky residue, which was purified by flash chromatography on silica (hexane:CH2Cl2 1:1) to afford a sticky orange solid. Recrystallisation from CH2Cl2/hexane gave H3 as an orange solid (0.77 g, 65%), m.p. 101–103 °C; 1H NMR showed the product was a mixture of diastereomers with estimated ratio 4:1. δH (300 MHz, CDCl3): 3.85 (1H, m, 1′′-H), 3.67 (0.2H, m, 5-H), 3.54 (0.8H, m, 5-H), 3.29 (4H, m, 5′-,6′-H2), 2.11 (0.2H, d, J = 5.4 Hz, OH), 2.00 (0.8H, d, J = 3.6 Hz, OH), 1.58 (4H, m, 2′′-,3′′-H2), 0.95 (3H, t, J = 7.0 Hz, 4′-H3); δC (75 MHz, CDCl3): 115.05, 114.35, 113.87, 111.89 (6 × sp2C), 72.41 & 72.25 (1′′-C), 52.77 & 50.25 (5-C), 36.76 & 36.56 (2′′-C), 33.18 & 31.54 (6-C), 30.20 (5′-,6′-C), 18.92 & 18.83 (3′′-C), 13.97 & 13.95 (4′′-C); IR νmax (cm−1, KBr): 3402 (br), 3334 (br, sh), 2951 (s), 2918 (s), 2864 (m), 1655 (w), 1514 (w), 1456 (w), 1408 (m), 1282 (m), 1223 (w), 1115 (m), 1066 (m), 1020 (m), 1003 (m), 908 (s), 847 (w), 770 (s), 592 (w), 503 (w), 449 (w); m/z: (FAB) 456 ([M]+, 100%); elem. anal. found C: 36.89, H: 3.54%; C14H16OS8 requires C: 36.84, H: 3.51%.
Preparation of [(H1)I3]·0.5I2 (18)
A solution of donor H1 (12 mg) in CH2Cl2 (3 mL) was gently added to a solution of iodine (8 mg) in CH2Cl2 (3 mL). Slow evaporation in the dark afforded a few black crystals after one week, m.p. 195 °C (dec); elem. anal. found C: 14.38, H: 1.13%; C11H10I4OS8 requires C: 14.32, H: 1.09%.Preparation of [(HMET)I3] (19)
A solution of iodine (20.1 mg) in acetonitrile (8 mL) was gently layered on top of a solution of HMET (12.5 mg) in THF (6 mL) in a test tube and left to stand in the dark for 1 week affording black plate-like crystals, m.p. 190 °C (dec); IR νmax (cm−1, KBr): 3280 (w), 2911 (w), 2170 (w), 1592 (m), 1450 (s), 1397 (s), 1332 (s), 1281 (m), 1141 (m), 1118 (m), 1047 (m), 996 (s), 889 (s), 766 (m), 743 (w); HRMS: (EI) found [M]+ 413.8488, C11H10OS8 requires 413.8492; elem. anal. found C: 16.95, H: 1.29; C11H10I3OS8 requires C: 17.20, H 1.23%.X-ray structure determination
Single crystals of H1, H2, 13, and the radical cation salt 18 were mounted on a cryoloop with paratone oil and examined on a Bruker APEX-II CCD diffractometer equipped with an Oxford Cryoflex low temperature device. Data were measured at 150(2) K using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) and the APEX-II software.16 Final cell constants were determined from full least squares refinement of all observed reflections. The data were corrected for absorption SADABS.17 Cell refinement and data-reduction were carried out by SAINT.16 For 19, the X-ray data was collected on single crystals at 150 K on an Agilent Xcalibur diffractometer equipped with a Sapphire detector and an Oxford Cryosystems Cryocool low temperature device using the CrysAlis-Pro software package.18a With the exception of 13 and 19, the structures were solved by direct methods (SHELXS-97) and refined with full least squares refinement on F2 using SHELXL-97 within the Bruker SHELXTL suite.18b One of the six membered rings in H1 is disordered and was modelled over two sites. Discorded solvent was removed from the final model of H1 using PLATON SQUEEZE.19 Compounds 13 and 19 were solved with direct methods (SHELXS-97) and refined using SHELXL-2013.18a The structure of 13 exhibited disorder of cyclopentane ring that was modelled over two sites. Initial structure solution of 19 revealed 1.5 BEDT fragments in the asymmetric unit and one well-ordered I3− anion. However the second I3− ion was severely disordered over multiple sites about a special position. A total of 9 positions were modelled with a total site occupancy of 1.5 which brought the residual electron density within 1.5e− Å−3. Attempts to clearly identify the CH2OH group positions on each BEDT core were unsuccessful although some low intensity peaks in the difference map were evident suggestive of severe disorder in the positions of the CH2OH units. The unsaturated nature of the backbone afforded 8 possible positions for the CH2OH group (and four for the CH2OH group attached to the BEDT molecule located about a special position). With a shortage of well-defined potential positions in the difference map, CH2OH side-chains were added using a FRAG⋯FEND command. Their positions were subsequently refined using appropriate 1,2- and 1,3-distance restraints (DFIX) and their site occupancies refined using a common thermal parameter (EADP). For the BEDT fragment on a general position four CH2OH positions were modelled, one on each of the crystallographically independent unsaturated C atoms of the BEDT backbone to afford a total site occupancy of 1.0 (SUMP). For the BEDT molecule located about a special position an additional two CH2OH groups were modelled with a total site occupancy of 0.5 (SUMP). Notwithstanding these efforts the Uiso for both the C and O atoms of the CH2OH groups were abnormally large indicating that the disorder was even more severe than the current model indicates. Attempts to model the disorder over even more sites was not considered. Crystallographic parameters for H1, H3, 13, 18 and 19 are summarized in Table 1.†| Complex | H1 | H2 | 13 | 18 | 19 |
|---|---|---|---|---|---|
| Formula | C11H10OS8 | C16H16O4S8 | C19H20O4S8 | C22H2OI6.6O2S16 | C11H10OS8I3 |
| Formula mass | 414.67 | 528.77 | 568.83 | 1666.37 | 795.37 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Crystal size/mm | 0.32 × 0.10 × 0.08 | 0.60 × 0.50 × 0.40 | 0.30 × 0.23 × 0.05 | 0.3 × 0.1 × 0.01 | 0.26 × 0.20 × 0.04 |
| Description | Plate | Block | Plate | Plate | Plate |
| Crystal colour | Orange | Orange | Orange | Black | Black |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P |
P |
I2/c |
| a/Å | 6.5765(10) | 14.2616(10) | 6.5807(3) | 7.9246(4) | 14.5595(7) |
| b/Å | 15.314(2) | 11.4272(8) | 12.1870(7) | 8.6449(4) | 13.7315(7) |
| c/Å | 16.664(2) | 12.9575(9) | 15.5413(9) | 16.2901(8) | 33.707(3) |
| α/° | 89.607(7) | 90 | 111.004(3) | 87.891(2) | 90 |
| β/° | 89.350(8) | 95.606(3) | 100.434(3) | 86.545(2) | 98.022(6) |
| γ/° | 88.861(7) | 90 | 91.420(3) | 76.982(2) | 90 |
| V (Å3) | 1677.8(4) | 2101.6(3) | 1138.86(11) | 1085.02(9) | 6672.9(8) |
| Temp./K | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| Z | 4 | 4 | 2 | 1 | 12 |
| Reflns collected | 45625 |
65807 |
50303 |
42831 |
19151 |
| Unique reflns | 10176 |
10180 |
5642 | 3808 | 5873 |
| Rint | 0.073 | 0.025 | 0.063 | 0.0696 | 0.0608 |
| R1 (I > 2σ(I)) | 0.043 | 0.032 | 0.029 | 0.0696 | 0.0893 |
| wR2 (all data) | 0.116 | 0.079 | 0.078 | 0.1872 | 0.2140 |
| Goof | 1.06 | 1.09 | 1.04 | 1.05 | 1.115 |
| Number of parameters | 386 | 255 | 301 | 232 | 313 |
| ΔρmaxΔρmin (e Å−3) | +0.71, −0.64 | +1.12, −0.45 | +0.58, −0.35 | +3.96, −2.56 | +1.29, −1.24 |
Computational studies
DFT calculations were carried out on H1 and H2 with initial geometry optimizations undertaken using the Pople 6-31G*+ basis set and B3LYP functional within Jaguar.20 Subsequent single-point energy calculations were performed on the optimized structure using the larger triple zeta 6-311G-3DF-3PD basis set.21Thin film preparation
:1 weight ratio of donor/PS on a Si wafer substrate was prepared by spin coating a 2 wt% solution of donor/PS (1:1 weight ratio) in chlorobenzene at 1000 rpm for 45 s. The film was dried in a vacuum oven at 65 °C for 3h to remove any solvent residue and then Au electrodes were deposited onto the film via vacuum deposition through a shadow mask. The film was then doped with iodine vapour by suspending the film above an I2/CH2Cl2 (100 mg/20 mL) solution for 8 min at RT.000) (20 μm thickness) on a glass substrate containing a 2 wt% of molecularly dispersed donor in a polystyrene matrix were obtained by casting from a solution of PS polymer and BEDT-TTF derivative in o-dichlorobenezene at 120 °C. The film was then exposed to iodine vapour by suspending the film over an I2/CH2Cl2 solution (0.1 g/20 mL) for 8 min. Such treatment resulted in the formation of a continuous network containing (H3)x-(polyiodides)y in a surface layer of the polystyrene film.Results and discussion
Synthesis
Building on our previous studies,13,14 a family of four hydroxyl functionalized BEDT-TTF derivatives were prepared, comprising three new donors H1–H3 as well as the previously reported HMET 2.13d H1 and H2 are achiral, HMET is a racemate and H3 was isolated as mixture of two diastereoisomers. It should be noted that for a side chain located on the six-membered ring of a BEDT-TTF derivative, the conformation of the ring can adjust between half chairs so that the R or S configured side chain can be orientated into similar positions. The general synthetic strategy for the preparation of the donors involves preparation of fused 1,3-dithiole-2-thiones, protection of their hydroxyl groups as acetates followed by formation of the central carbon–carbon double bond via a phosphite-mediated coupling reaction of the thione or its corresponding oxo compound and subsequent deprotection of the hydroxyl group. Thus, for H1 which has terminal six- and seven-membered rings, the hydroxyl substituted seven-membered ring thione 3 was obtained by cyclisation of the zinc complex of 2-thioxo-1,3-dithiole-4,5-dithiolate, (NEt4)2[Zn(dmit)2]26,27 with 1,3-dibromopropan-2-ol, Scheme 1.28 The hydroxyl group was protected as an acetate to give 4 which was converted to the corresponding oxo compound 5 by treatment with mercuric acetate. Reaction of 5 with the unsubstituted six-membered ring thione 629 in triethyl phosphite afforded the cross-coupled acetyl-protected donor 7 in 66% yield, along with BEDT-TTF and a small amount of the homo-coupled bis-acetyl protected donor 8 (5%) after chromatography. Deprotection of 7 with HCl in THF afforded H1 in an almost quantitative yield. | ||
| Scheme 1 | ||
The bis(hydroxymethyl) donor H2 was prepared via the cycloaddition of 3,3-bis(hydroxymethyl)cyclopentene30 with trithione 9 to give thione 10 in 66% yield which was converted to the bis(acetate) 11 and then treated with Hg(OAc)2 to provide the oxo compound 12 in 86% yield.
Cross-coupling of 12 with the unsubstituted thione 6 afforded donor 13 in 53% yield. Deprotection of 13 in acidic conditions afforded donor H2 as a yellow solid in 65% yield, Scheme 2.
 | ||
| Scheme 2 | ||
Donor H3, a BEDT-TTF derivative bearing a 1-hydroxybutyl side chain, was prepared from trithione 929 via cycloaddition with hex-1-en-3-ol to give thione 14 as a (65:35) mixture of diastereomers in 71% yield. Protection of the hydroxyl group as acetate 15, conversion of this thione to the corresponding oxo derivative 16 and cross coupling with the unsubstituted thione 6 afforded the mono-substituted donor 17 in 56% yield together with the homo coupled adducts. Deprotection of 17 with hydrochloric acid in THF afforded donor H3 as a mixture of diastereomers which could not be completely separated by chromatography or recrystallization, Scheme 3.
 | ||
| Scheme 3 | ||
X-ray crystallography
Single crystals of H1 and H2 and the O-acetylated precursor 13 were characterized by X-ray diffraction to study the dominant molecular interactions in the crystal packing of the neutral donors. Given that the molecular structures of these compounds dictate their electrical properties, crystallographic studies are crucial to shed light on how synthetic modifications to the BEDT-TTF framework disrupts the tendency of the donors to stack in the solid state. Finally, in order for these materials to become electronic conductors they are typically doped with iodine. In order to investigate how the crystal structures of the donors are altered after oxidation to radical cations, donors H1 and HMET were doped with iodine and the structures of their radical cation salts were elucidated by X-ray diffraction. For all five structures, selected bond lengths and angles are reported in S-2 of the ESI.†Hydroxyl donors H1 and H2
Single crystals of H1 were obtained via diffusion of hexane into a THF solution of the donor. The compound crystallizes in the triclinic space group P with two independent molecules, A and B, in the asymmetric unit, Fig. 2.
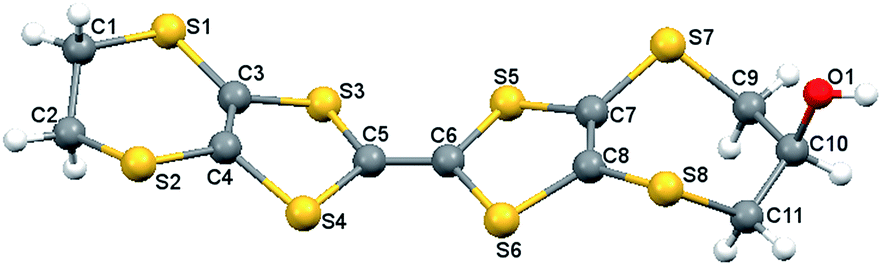 | ||
| Fig. 2 Molecular structure of one independent molecule (B) of H1 with atomic labelling scheme. | ||
Both donors are slightly bowed due to bends about their dithiole S⋯S vectors in the range 17.9–23.9°. The ethylene bridges in both molecules are disordered between two half chair conformations for molecule A and two envelope conformations for molecule B, a feature which is common for such systems.13,14 The seven-membered rings of both molecules crystallize in chair conformations with the hydroxyl groups adopting pseudo-axial orientations. The molecules are packed as centrosymmetric pairs in a head-to-tail manner with neighbouring dimers situated almost at right angles to one another, Fig. 3.
 | ||
| Fig. 3 Crystal packing arrangement of H1. View down the a*-axis showing four dimers participating in a hydrogen bonded “square motif.” S⋯S contacts between pairs of dimers and H-bonding interactions between hydroxyl groups are shown as red dashed lines. H-atoms are omitted for clarity. | ||
Within each pair, the central TTF units are offset from one another along the molecular axis so there are two S⋯S contacts per dimer of 3.5067(8) and 3.6065(8) Å, close to the S⋯S van der Waals distance of 3.6 Å. Donor pairs are assembled into layers in the ca + b plane, such that the hydroxyl groups of four donors can hydrogen bond to each other forming a roughly square arrangement between the oxygen atoms (O⋯O: 2.673(2) and 2.727(2) Å), Fig. 3. This type of square shaped hydrogen bonded motif has also been observed previously for the triiodide salt of bis(2R-2,3-dihydroxypropylthio) (ethylenedithio)TTF.13b For H1 the hydrogen bonding between donors dominates the crystal packing, and there are no short S⋯S contacts between co-parallel stacks of donors in the crystal lattice. In contrast, there are several short edge-to-edge S⋯S contacts between donors in neighbouring layers of 3.4121(8) Å and several longer interactions in the range 3.5067(8)–3.594(1) Å that could mediate electronic communication between donor molecules, Fig. 4. Channels running through the structure contain disordered hexane molecules which were excluded from the final model using PLATON SQUEEZE.19
 | ||
| Fig. 4 Crystal packing arrangement of H1, showing S⋯S contacts (red dashed lines) between molecules belonging to adjacent donor pairs. H-atoms are omitted for clarity. | ||
Single crystals of H2 were obtained by the diffusion of Et2O into a DMF solution of the donor. The molecule crystallizes in the monoclinic space group P21/c. The framework of the donor is slightly bowed due to small bends of 12.9 and 13.4° about the dithiole S⋯S vectors. The fused cyclopentane ring adopts a half chair conformation with the largest torsion about a CH–CH2 bond, Fig. 5.
 | ||
| Fig. 5 Molecular structure of H2 with atomic labelling scheme. | ||
The donors are hydrogen bonded into layers which lie at 63° to the c-axis with four donors contributing one hydroxyl group to a hydrogen bonded square motif of oxygen atoms, similar to that observed in the structure of H1, Fig. 6a.
 | ||
| Fig. 6 (a) Crystal packing arrangement of molecules of H2 into layers showing S⋯S and H-bonding interactions as red dashed lines; (b) view down the b-axis with the donor layers running horizontally. H-atoms are omitted for clarity. | ||
The O⋯O distances are 2.701(7) and 2.762(8) Å, and there are short S⋯S contacts of 3.323(3), 3.523(3) and 3.525(3) Å between donors. The unit cell contains eight successive layers, with no π-stacking of donors and just a few short S⋯S contacts in the range of 3.562(3) to 3.737(3) Å, Fig. 6b. Unfortunately, establishing suitable conditions for the growth of suitable single crystals from a diastereoisomeric mixture proved particularly challenging and thus no suitable crystals of H3 have been obtained to date.
O-Acetyl-protected donor 13
The crystal structure of the O-acetyl-protected 13 was determined to assess how the larger acetyl substituents as well the absence of the OH groups affect the packing arrangement of the BEDT-TTF derivatives. Single crystals of 13 were obtained via slow diffusion of diethyl ether into a CH2Cl2 solution of the protected donor.The donor crystallises in the triclinic space group P with two independent molecules in the asymmetric unit. In contrast to the bowed geometries of the previously described donors, this molecule adopts a planar configuration for the central organosulfur residue with only small torsion angles about the two dithiole S⋯S vectors of 4.8 and 5.3°, Fig. 7. The flexibility about these S⋯S vectors is a feature of this class of molecules in their neutral states. The molecules are packed in centrosymmetric pairs, with a dithiole group of one donor lying over the central double bond of the other and vice versa, which allows a pair of splayed acetyl groups to extend beyond each end of the pair. The pairs are packed in columns in the bc plane with the long molecular axes at ca. 45° to the b-axis and significantly offset to accommodate the two acetate groups resulting in no short π-stacking interactions, Fig. 8a. However, there are four short edge-to-edge S⋯S contacts between donors in adjacent columns in the range of 3.3772(6)–3.5881(5) Å, Fig. 8b. For comparison, a summary of the S⋯S contacts and H-bonding interactions in all five structures is presented in Table 2.
 | ||
| Fig. 7 Molecular structure of 13 with atomic labelling scheme. | ||
 | ||
| Fig. 8 (a) Crystal packing of centrosymmetrically related pairs of molecules of 13; (b) S⋯S contacts between adjacent columns of 13 shown as red dashed lines. H-atoms are omitted for clarity. | ||
| Donor | Dimer S⋯S (Å) | Edge S⋯S (Å) | D⋯A (Å) | S⋯I (Å) |
|---|---|---|---|---|
| H1 | 3.5067(8) | 3.4121(8) | 2.673(2) | — |
| 3.6065(8) | 3.5067(8) to 3.595(1) | 3.727(2) | ||
| H2 | — | 3.323(3) | 2.701(7) | — |
| 3.523(3) | 2.762(8) | |||
| 3.525(3) | ||||
| 13 | — | 3.3772(6) to 3.5881(5) | — | — |
| 18 | 3.352(4) | 3.572(4) | 2.87(2) | 3.639(5) to 3.648(2) |
| 3.462(4) | ||||
| 3.592(5) | ||||
| 19 | 3.365(5) | 3.427(6) | 3.604(4) to 3.742(5) | |
| 3.394(6) | ||||
| 3.667(6) |
Attempts were made to oxidize the donors with iodine and to characterize the molecular structures of the resulting charge transfer salts by X-ray crystallography. Following this strategy we were successful in finding suitable conditions for the growth of suitable single crystals of two radical cation salts, 18 and 19.
Radical cation salts [(H1)+˙I3−]·0.5I2 (18) and (HMET)+˙I3− (19)
BEDT-TTF forms radical cation salts readily with iodine, affording a number of different stoichiometries and polymorphs that all contain triiodide anions and in some cases also neutral iodine molecules.31 Previous studies have revealed that the triiodide salts of hydroxyl-substituted donors tend to have 1:1 stoichiometries with the “soft” triiodide anions preferring to lie closer to sulfur atoms than to hydroxyl groups.13b,32 Single crystals of 18 were prepared via the slow diffusion of an iodine solution into a solution of H1 in dichloromethane. X-ray crystallography reveals that in contrast to the molecular structure of the neutral donor, there is one unique radical cation in the asymmetric unit whose 7-membered ring adopts a chair conformation, Fig. 9. After oxidation, the BEDT-TTF derivatives adopt a more planar conformation which can be clearly seen when comparing the molecular structures of donor H1 before and after oxidation, Fig. 10 and Table 3.
 | ||
| Fig. 9 Molecular structure of [H1]+˙ with atomic labelling scheme. H-atoms are omitted for clarity. | ||
 | ||
| Fig. 10 The molecular structure of donor H1 (a) when neutral and (b) after oxidation to the radical cation. H-atoms are omitted for clarity. | ||
C and C–S bond lengths in H1 and [H1]˙+ in 18
| H1 | [H1]˙+ | ||
|---|---|---|---|
| Bond | Distance (Å) | Bond | Distance (Å) |
| C5–C6 | 1.345(3) | C16–C17 | 1.387(14) |
| C6–S5 | 1.7571(19) | C16–S13 | 1.721(9) |
| C6–S6 | 1.7578(19) | C16–S14 | 1.713(9) |
| C5–S3 | 1.758(2) | C17–S15 | 1.727(9) |
| C5–S4 | 1.7607(19) | C17–S16 | 1.728(9) |
| S5–C7 | 1.7609(19) | S13–C14 | 1.732(11) |
| S6–C8 | 1.7611(19) | S14–C15 | 1.735(9) |
| S3–C3 | 1.761(2) | S15–C18 | 1.742(9) |
| S4–C4 | 1.776(2) | S16–C19 | 1.737(9) |
| C7–C8 | 1.341(3) | C14–C15 | 1.361(13) |
| C3–C4 | 1.345(3) | C18–C19 | 1.359(13) |
The C–S bond lengths (1.721(9)–1.742(9) Å) and CC bond lengths (C14C15, C16C17, C18C19 are 1.361(13), 1.387(14) and 1.359(13) Å, respectively) are in good agreement with those observed in other BEDT-TTF+˙ charge transfer salts.33 For clarity, selected bond lengths of molecule B of the neutral donor H1 and its oxidized counterpart [H1]+˙ in 18 are compared in Table 3. As expected, the C–S bond lengths of the donor decrease upon oxidation by ca. 0.035 Å and the central CC bond shows the most change, being lengthened by ca. 0.04 Å.33b
The crystals of 18 are comprised of centrosymmetric pairs of donor radical cations oriented in a head-to-tail manner surrounded by iodine molecules and a disordered array of triiodide ions corresponding to a formula of [(H1)I3]·0.5I2 (18) that is further supported by the elemental analysis data, Fig. 11. The disordered triiodide anions fill the space between iodine molecules in the c-direction between adjacent cells, and also fill channels running in the a-direction throughout the crystal. The organosulfur heterocycles of the cation pair lie opposite each other with eight S⋯S contacts; two each of 3.352(4), 3.365(4) and 3.462(4) Å and a longer one of 3.592(5) Å, Fig. 12a (red dashed lines). S⋯S contacts of 3.572(4) Å between dimer pairs are shown as blue dashed lines in Fig. 12a.
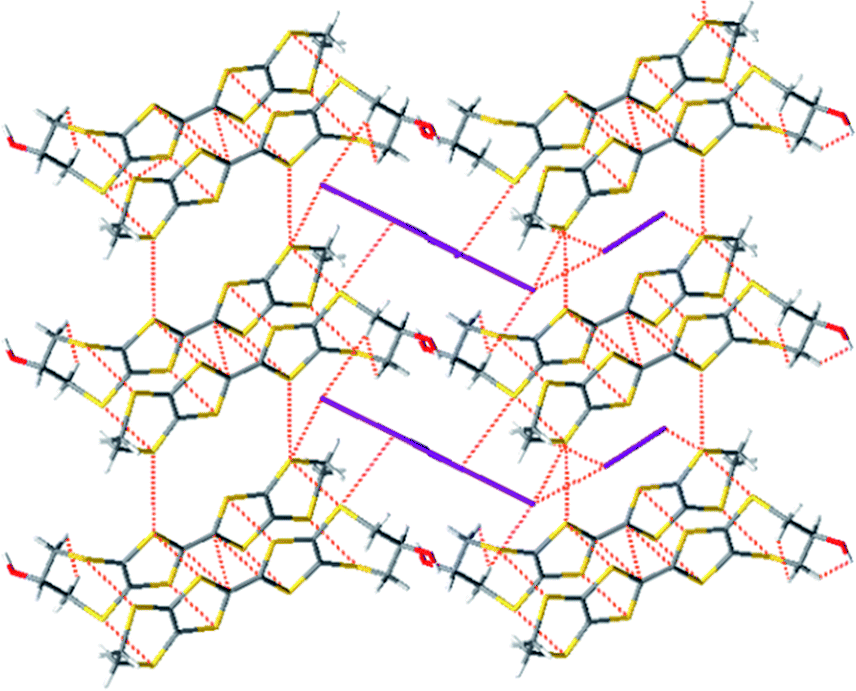 | ||
| Fig. 11 Crystal packing of 18 showing the head-to-tail arrangement of donor cations in dimers that form channels which accommodate the I3− counterions and neutral I2 molecules. | ||
 | ||
| Fig. 12 (a) Crystal packing of 18; short S⋯S contacts between dimers are shown in red; S⋯I contacts are shown in green and inter-dimer S⋯S contacts are shown in blue. (b) Crystal packing of the oxidized donors with the disordered triiodides removed showing the neutral iodine molecules organized in the channels between stacks of dimers. H-atoms are omitted for clarity. | ||
The iodine molecules lie between the donor cation pairs with S⋯I contacts in the range of 3.639(5)–3.648(2) Å, Fig. 12b. In contrast to the crystal structure of the neutral donor there are no H-bonded square motifs, but there are hydrogen bonds between the cation pairs located “end to end” (O⋯O = 2.87(2) Å).
Single crystals of the radical cation salt of HMET 19, were grown via the slow diffusion of an acetonitrile solution of iodine into a THF solution of the donor. The compound crystallizes in the monoclinic space group I2/c with 1.5 independent donors and 1.5 tri-iodide anions in the asymmetric unit. The molecular structure of the donors is shown in Fig. 13.
 | ||
| Fig. 13 Molecular structure of the two independent molecules of [HMET]+˙ in 19 with the appropriate atomic labelling scheme. For clarity, only one orientation of the disordered side chains is shown. H-atoms are omitted for clarity. | ||
Once again the donors adopt a more planar conformation after oxidation, with the first donor packing as part of a centrosymmetric pair stabilized by four sets of face-to-face S⋯S contacts between 3.365(5) and 3.667(6) Å, Table 2. The second donor acts as a spacer where its sulfur atoms participate in edge-to-edge contacts (3.427(6) Å), connecting it to the dimers both above and below it in a stack that propagates along the b-axis, Fig. 14.
 | ||
| Fig. 14 View down the c-axis showing one stack of HMET donors in 19. S⋯S contacts are shown as red dashed lines. H-atoms are omitted for clarity. | ||
Further examination of the crystal packing reveals that the stacks are interspersed by two sets of tri-iodide counter anions. The first set occupies the spaces between the donor molecules and the second run perpendicular to the stacks of donors along the a-axis, Fig. 15. The former anion is well-located but the second I3− anion is poorly located indicative of some static or dynamic disorder. As is common for these systems, the packing arrangement is further stabilized by a series of short S⋯I contacts that range from 3.604(4) to 3.742(5) Å, Fig. 15. Interestingly, this arrangement is different from crystal packing of the HMET radical cation in the charge transfer salt (HMET)[Cr(NAOP)(NCS)2] (NAOP2− = N,N′-(1,2-phenylenebis(nitrilomethylidyne))bis(2-naphthoate)).33
 | ||
| Fig. 15 Crystal packing of [HMET]+˙I3−. View down the b-axis showing the arrangement of the donors and the iodide anions. S⋯S and S⋯I contacts are shown as red dashed lines. The disordered hydroxymethyl side chains of the donors and the H-atoms are omitted for clarity. | ||
In the latter case, the radical cations are organized into pairs that are isolated from each other, but this may in part be due to the bulky nature of the complex anions in the crystal structure. Further comparison of the two HMET charge transfer salts reveals that for both compounds the hydroxylmethyl side chains are extremely disordered making the refinement of their crystal structures problematic. This may also explain why suitable conditions for the growth of single crystals of the neutral HMET donor have proven elusive to date. More importantly, this also reveals that in certain cases unsymmetrically substituted donors struggle to pack efficiently in the solid state, which may have a negative impact on their electron mobilities. In order to investigate this further we investigated the electronic properties of this family of donors both in solution and after fabrication into polymeric thin films.
Electrochemical properties
The redox potentials of H1–H3 and HMET 2 were investigated by cyclic voltammetry. The results are summarized in Table 4, together with that of BEDT-TTF for comparison.| Compound | E11/2/V | E21/2/V | ||
|---|---|---|---|---|
| THF | CH2Cl2 | THF | CH2Cl2 | |
| BEDT-TTF | 0.68 | 0.48 | 0.83 | 0.89 |
| H1 | 0.70 | 0.51 | 0.86 | 0.91 |
| H2 | 0.72 | — | 0.87 | — |
| H3 | 0.74 | 0.53 | 0.90 | 094 |
| HMET 2 | 0.69 | 0.51 | 0.85 | 0.91 |
For donor H2 which had poor solubility in CH2Cl2, the redox properties were measured in THF. All donors show two reversible single-electron redox processes assigned to the formation of the radical cation and dication respectively. The calculated HOMO densities of H1 and H2 are similar. The HOMO density is high on the sulfur atoms of the TTF core, but negligible on the sulfur atoms of the outer rings as well as their substituents (Fig. 16).
 | ||
| Fig. 16 The HOMO coefficients on H1 and H2 (DFT B3LYP/6-31G(d)).20,21 The HOMO energies are calculated to be −4.93 and −4.95 eV respectively. | ||
Comparing the energies of the two HOMOs it is apparent that H1 is slightly higher in energy which is consistent with the electrochemistry data suggesting that it might be slightly easier to oxidize, Fig. 16.
Thin film studies on H3
Given the compromised solubilities of H1 and H2 in comparison with HMET 2, we proposed to introduce a longer alkyl side chain into the BEDT-TTF derivative to target the preparation of a more soluble donor for fabrication into thin films. As expected, the particularly good solubility of H3 in organic solvents made this donor an ideal candidate to establish synthetic protocols for the preparation and study of polymer-blend thin films. Two sets of thin films of H3 with a thickness of ∼27 nm were first prepared on (i) unmodified and (ii) OTS modified Si wafer substrates. The films were then doped with iodine vapour from an I2/CH2Cl2 solution. The specific conductivities and resistivities of these films were then determined and the results are summarized in Table 5. Interestingly, for the doped film of H3 on unmodified substrate, the conductivity was 3 times higher than the 2% composite films prepared from the thiophene appended BEDT-TTF donor 1.14| Material | Specific conductivity S cm−1 | Resistivity Ω cm |
|---|---|---|
| H3/PS OTS modified Si substrate, undoped | 3.7 × 10−8 | 2.7 × 107 |
| H3/PS modified OTS Si substrate I2 doped | 1.1 × 10−4 | 9 × 103 |
| H3/PS unmodified Si substrate, undoped | 5.0 × 10−8 | 2.0 × 107 |
| H3/PS unmodified OTS Si substrate I2 doped | 2.2 × 10−5 | 4.5 × 104 |
| H3/2% PS glass substrate I2 doped | 5 × 10−3 | 200 |
| Thiophene donor 1/2% PS glass substrate I2 doped14 | 3.5 × 10−5 | 2.8 × 104 |
FET characterization of doped films comprised of a 1:1 weight ratio of H3 relative to polystyrene (PS) deposited on an unmodified silica wafer substrate showed IDS–VDS curves of ohmic and linear properties consistent with quasiconducting behaviour and all the I–V curves overlapped with each other when the gate voltage was changed from +10 to −40 V in 10 V intervals (Fig. 17a).
 | ||
| Fig. 17 Drain current versus drain voltage as a function of gate voltage for a device based on (1:1) donor/PS blend films on unmodified Si wafer substrate at room temperature. (a) I2-doped H3/PS film; (b) undoped H3/PS film (the gate voltage was changed from 10 to −40 V in 10 V intervals). | ||
In contrast, un-doped films of H3/PS gave rise to the IDS–VDS plots with profiles shown in Fig. 17b, consistent with an insulating material. From these results we can conclude that iodine vapour doping of the polymer blend film of the donors on Si wafer substrates produces continuous conducting-like layers. A device was then fabricated from a thin film of H3 as follows: an n-doped Si wafer with a 110 nm thermally grown silicon dioxide layer (capacitance of 32 nF cm−2) was used as the substrate. Semiconductor films were then deposited by spin coating 1 wt% of chlorobenzene solution of the donor onto octyltrichlorosilane (OTS) modified and unmodified substrates, respectively, at 1000 rpm for 45 s. Gold source and drain electrodes were then deposited over the substrate by vacuum deposition methods through a shadow mask with various channel lengths and widths, Fig. 18.
 | ||
| Fig. 18 Schematic diagram of the OTFT device configuration. | ||
Unfortunately, no field effect was observed for the device which we propose is either due to (i) structural defects in the polycrystalline film at least in part due to a disorder of the side chains as observed in the radical cation salts of the closely related HMET donors, or (ii) to a charge trapping effect mediated by the hydroxyl substituents of the donors which is already known to seriously compromise the OFET properties of films fabricated from hydroxyl functionalized polymers.34
In order to study the electronic properties of the polymer blend thin-films by Vis-NIR spectroscopy, a surface conducting polystyrene (PS) film deposited on a glass substrate was prepared applying a two-step reticulate doping technique.24,25,35 Films comprising 2 wt% of H3 donor in a PS composite were exposed to iodine vapour from a I2/CH2Cl2 solution (0.1 g/20 mL). Such treatment results in the deposition of a continuous network containing (H3)x-(polyiodides)y onto the surface layer of the polystyrene films.14 It should be noted that since the real thickness of the conducting layer is unknown the exact resistivity and specific conductivity of the network in the films cannot be determined. The un-doped film exhibited insulating behavior with conductivities comparable to the glass substrate. For the doped film, if we assume the conducting layer thickness to be approximately the same as the thickness of the polymer film (20 μm), a resistivity (ρ) of 200 Ω cm and conductivity (σ) of 5 × 10−3 S cm−1 was estimated, suggesting that there is an efficient pathway for electron mobility. We also can A comparison of Vis-NIR spectra for doped and undoped films of H3 in a polystyrene matrix is shown in Fig. 19.
 | ||
| Fig. 19 Vis-NIR spectra of un-doped and doped films on glass substrates for a 2 wt% ratio of H3 relative to PS. | ||
The absorption bands at 950 and 3000 nm are assigned to mixed-valence CT states and are consistent with the realization of a π-conducting network that facilitates conductivity.36 In contrast to the recently characterized PS composites of thiophene appended BEDT-TTF derivatives, no broad absorption band between 1250 and 1500 nm was observed, ruling out the presence of any fully oxidized salts in the polymer matrix.14
Conclusion
We have prepared and characterized three new hydroxyl-substituted BEDT-TTF derivatives, together with an acetyl intermediate and two radical cation salts. Our studies reveal (i) several types of short contacts including H-bonding. S⋯S and S⋯I interactions organize the donors and acceptors in the solid state and that the importance of hydrogen bonding in organizing the donors in their neutral state is overridden by S⋯S and S⋯I interactions, which are likely responsible for the onset of conductivity on doping; (ii) hydroxyl groups improve the electron transport properties of the thin films deposited on glass substrates and (iii) the unsymmetrical derivatives are susceptible to disorder in the solid state which may compromise their electron transport properties. Donor H3 was successfully incorporated into two sets of polymeric thin films deposited on either silica or glass substrates. Both types of films displayed quasi-conducting properties upon doping with I2. The lack of any OFET response for devices fabricated from thin films deposited onto silica can be primarily attributed to the hydroxyl substituents of the donors combining with water molecules acting to trap the charges at the interface of the device between the semiconductor and the dielectric media. This phenomenon is not unique to this system; trap states in organic semiconductors are currently the subject of intense study and are known to severely compromise the performance of OFET devices.34 Although the hydroxyl donors studied here are clearly not suitable candidates for organic field effect transistors, they do afford thin films whose electronic properties and solubilities can be chemically tuned and thus may yet lend themselves to applications within the field of organic electronics.Acknowledgements
Financial support from NSERC (Discovery, MP and JMR; Strategic Supplemental, MP), CRC (Tier II MP; Tier I JMR), CFI and ORF (New Opportunities, MP), Brock University (International Seed Funds MP) (Ministry of Ontario, Early Researchers Award, MP). Nottingham Trent University is gratefully acknowledged for a sabbatical period (JDW). JDW thanks the EPSRC UK National Mass Spectrometry Facility at Swansea University for data. We also gratefully acknowledge the XRCC for access to facilities and expertise to carry out the thin film and OFET part of this study.References
- (a) F. Wudl, G. M. Smith and E. J. Hufnael, J. Chem. Soc., Chem. Commun., 1970, 1453 RSC; (b) S. Hunig, G. Kiesslich, D. Scheutzow, D. Zharadnik and P. Carsky, Int. J. Sulfur Chem., 1971,(part C), 109 Search PubMed; (c) M. Nazario, Chem. Commun., 2013, 49, 7025 RSC.
- (a) M. R. Bryce, J. Mater. Chem., 2000, 10, 589 RSC; (b) J. L. Segura and N. Martın, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS; (c) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef CAS PubMed; (d) P. Batail, Chem. Rev., 2004, 104, 4887 CrossRef CAS; (e) E. Doni and J. A. Murphy, Chem. Commun., 2014, 50, 6073 RSC.
- (a) D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC; (b) V. A. Azov, J. Cordes, D. Schfeuler, T. Boeckmann, D. Marcus and L. Nikos, J. Org. Chem., 2014, 79, 1174 CrossRef PubMed; (c) T. Avellini, H. Li, A. Coskun, E. Barin, A. Trabalsi, A. Basvray, N. Ashish, K. Sanjeev, A. Credi, S. Silvi and F. J. Stoddart, Angew. Chem., Int. Ed., 2012, 51, 1611 CrossRef CAS PubMed; (d) C. Simao, M. Mas Torrent, J. Casado-Montenegro, F. Oton, J. Vecciana and C. Rovira, J. Am. Chem. Soc., 2011, 133, 13256 CrossRef CAS PubMed; (e) C. P. Collier, E. W. Wong, M. Belohradsky, F. M. Raymo, J. F. Stoddart, P. J. Kuekes, R. S. Williams and J. R. Heath, Science, 1999, 285, 391 CrossRef CAS.
- (a) J. Xiang, L. Long, W. Liu, J. E. Boves, Z. Yu-Xang and J.-L. Zuo, Tetrahedron Lett., 2013, 54, 1998 CrossRef PubMed; (b) M. Falk, V. Andoralov, M. Silow, M. D. Toscano and S. Shleev, Anal. Chem., 2013, 85, 6342 CrossRef CAS PubMed; (c) G. Yang, Z. Chong-an, G. Zhang, J. Zhang, J. Xiang, D. Zhang and D. Zhu, Adv. Funct. Mater., 2013, 23, 1671 CrossRef CAS PubMed; (d) M. Hardouin-Lerouge, B. Chesneau, M. Allain and P. Hudhomme, J. Org. Chem., 2012, 77, 2441 CrossRef CAS PubMed; (e) W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC.
- (a) F. Pop, S. Laroussi, T. Cauchy, C. Gomez-Garcia, J. Carlos, J. D. Wallis and N. Avarvari, Chirality, 2013, 25, 466 CrossRef CAS PubMed; (b) H. Muller, S. Fiedler, M. Saad and C. Riekel, Synth. Met., 1997, 86, 1885 CrossRef CAS; (c) T. L. Li and R. Giasson, J. Am. Chem. Soc., 1994, 11, 9890 CrossRef; (d) H. Akutsu, J.-I. Nakatsuji and S. S. Turner, Dalton Trans., 2013, 42, 16351 RSC; (e) L. Martin, P. Day, S. A. Barnett, D. A. Tocher, P. N. Horton and M. B. Hursthouse, CrystEngComm, 2008, 10, 192 RSC; (f) T. Enoki and A. Miyazaki, Chem. Rev., 2004, 104, 5449 CrossRef CAS PubMed.
- (a) E. Laukhina, R. Pfattner, L. R. Ferreras, S. Galli, M. Mas-Torrent, N. Masciocchi, V. Laukhin, C. Rovira and J. Veciana, Adv. Mater., 2010, 22, 977 CrossRef CAS PubMed; (b) E. Laukhina, R. Pfattner, M. Mas-Torrent, C. Rovira, J. Veciana and V. Laukhin, Sens. Transducers J., 2011, 10, 1 CAS; (c) H. Alves, A. S. Molinari, H. Xie and A. F. Morpurgo, Nat. Mater., 2008, 7, 574 CrossRef CAS PubMed; (d) R. M. Metzger, B. Chen, U. Hopfner, M. V. Lakshmikantham, D. Villaume, T. Kawai, X. Wu, H. Tachibana, T. V. Hughes, H. Sakurai, J. W. Baldwin, C. Hosch, M. P. Cava, L. Brehmer and G. J. Ashwell, J. Am. Chem. Soc., 1997, 119, 10455 CrossRef CAS; (e) D.-Y. Noh, A. G. Willing, Y. C. Han, K.-S. Shin, U. Geiser and H. H. Wang, Chem. Mater., 2004, 16, 4777 CrossRef CAS; (f) H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158 CrossRef CAS PubMed; (g) Organic Electronics: Materials, Manufacturing and Applications, ed. H. Klauk, VCH-Wiley, 2006 Search PubMed.
- (a) B. Mukherjee and M. Mukherjee, Langmuir, 2011, 27, 11246 CrossRef CAS PubMed; (b) R. Pfattner, M. Mas-Torrent, I. Bilotti, A. Brillante, S. Milita, F. Liscio, F. Biscarini, T. Marszalek, J. Ulanski and A. Nosal, Adv. Mater., 2010, 22, 4198 CrossRef CAS PubMed; (c) X. Gao, W. Qiu, Wenfeng, Y. Liu, G. Yu and D. Zhu, Pure Appl. Chem., 2008, 80, 2405 CrossRef CAS.
- (a) J. M. Williams, M. A. Beno, H. H. Wang, P. C. W. Leung, T. J. Emge, U. Geiser and K. D. Carlson, Acc. Chem. Res., 1985, 18, 261 CrossRef CAS; (b) P. Day, C. R. Chim., 2003, 6, 301 CrossRef CAS; (c) T. Mori, Bull. Chem. Soc. Jpn., 1998, 71, 2509 CrossRef CAS; (d) T. Mori, H. Mori and S. Tanaka, Bull. Chem. Soc. Jpn., 1999, 72, 179 CrossRef CAS; (e) T. Mori, Bull. Chem. Soc. Jpn., 1999, 72, 2011 CrossRef CAS; (f) T. Mori, Chem. Rev., 2004, 104, 4947 CrossRef CAS PubMed.
- (a) N. Yoshimoto, H. Maehara, Y. Ueda, J. Ni, K. Omote and M. Yoshizawa, Mol. Cryst. Liq. Cryst., 1998, 316, 129 CrossRef CAS PubMed; (b) S. Molas, J. Caroa, J. Santiso, A. Figueras, J. Fraxedas, C. Méziére, M. Fourmigué and P. Batail, J. Cryst. Growth, 2000, 218, 399 CrossRef CAS; (c) U. Geiser, H. H. Wang, C. Y. Han and G. A. Willing, NATO Sci. Ser., II, 2004, 139, 217 Search PubMed , Organic Conductors, Superconductors and Magnets, 231; (d) K. Kawabata, K. Tanaka and M. Mizutani, Solid State Commun., 1990, 75, 83 CrossRef; (e) M. Kilitziraki, A. J. Moore, M. C. Petty and M. R. Bryce, Thin Solid Films, 1998, 335, 209 CrossRef CAS.
- (a) V. I. Troitsky, T. S. Berzina, E. Dalcanale and M. P. Fontana, Thin Solid Films, 2002, 405, 276 CrossRef CAS; (b) V. I. Troitsky, E. Stussi, M. Mule and D. De Rossi, Synth. Met., 1993, 60, 111 CrossRef; (c) T. S. Berzina, S. A. Shikin, P. S. Sotnikov, V. I. Troitsky, V. Y. Khodorkovsky, O. Y. Neilands and G. Pukitis, Top. Mol. Organ. Eng., 1991, 7, 99 CAS.
- H. H. Wang, C. Y. Han, D.-Y. Noh, K.-S. Shin, G. A. Willing and U. Geiser, Synth. Met., 2003, 137, 1201 CrossRef CAS.
- H. X. Li, D. Q. Zhang, Y. Xu, W. Xu, G. Yu, X. G. Li and D. B. Zhu, Synth. Met., 2001, 123, 385 CrossRef CAS.
- (a) J.-P. Grifffiths and J. D. Wallis, J. Mater. Chem., 2005, 15, 347 RSC; (b) I. Awheda, S. J. Krivickas, S. Yang, L. Martin, M. A. Guziak, A. C. Brooks, F. Pelletier, M. Le Kerneau, P. Day, P. N. Horton, H. Akutsu and J. D. Wallis, Tetrahedron, 2013, 69, 8738 CrossRef CAS PubMed; (c) Q. Wang, P. Day, J. P. Griffiths, H. Nie and J. D. Wallis, New J. Chem., 2006, 1790 RSC; (d) N. Saygili, R. J. Brown, P. Day, R. Hoelzl, P. Kathirgamanathan, E. R. Mageean, T. Ozturk, M. Pilkington, M. M. B. Qayyum, S. S. Turner, L. Vorwerg and J. D. Wallis, Tetrahedron, 2001, 57, 5015 CrossRef CAS; (e) T. Ozturk, N. Saygili, S. Ozkara, M. Pilkington, C. R. Rice, D. A. Tranter, F. Turksy and J. D. Wallis, J. Chem. Soc., Perkin Trans. 1, 2001, 4, 407 RSC.
- Q. Wang, J. D. Wallis, Y. Wu and M. Pilkington, CrystEngComm, 2014, 16, 10235 RSC.
- A. Ueda, S. Yamada, I. Takayuki, H. Kamo, A. Akiko, R. Kumai, H. Nakao, Y. Murakami, K. Yamamoto and Y. Nishio, J. Am. Chem. Soc., 2014, 136, 12184 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (b) SAINT, v. 6.02, Bruker AXS Inc., Madison, WI, 1999 Search PubMed; (c) APEX-II, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- G. M. Sheldrick, SADABS, v. 2.05, University of Göttingen, Germany Search PubMed.
- (a) CrysAlisPro, Agilent Technologies, Version 1.171.35.15, release 03-08-2011; (b) SHELXTL package for crystal structure solution and refinementBruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- (a) A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–19 CrossRef CAS PubMed; (b) P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef; (c) A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, Netherlands, 2005 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- Jaguar version 8.0, Schrödinger LLC, New York, NY, 2013 Search PubMed.
- (a) M. Mas-Torrent, E. Laukhina, C. Rovira, J. Veciana, V. Tkacheva, L. Zorina and S. Khasanov, Adv. Funct. Mater., 2001, 11, 299 CrossRef CAS; (b) A. Tracz, J. K. Jeszka, A. Sroczyńska, J. Ulański and T. Pakula, Adv. Mater. Opt. Electron., 1996, 6, 335 CrossRef CAS.
- W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC.
- (a) P. Polanowski, A. Tracz, J. Ulański and E. Dormann, Synth. Met., 2000, 109, 235 CrossRef CAS; (b) J. K. Jeszka, A. Tracz, M. Sroczyńska, H. Kryszewski, S. Yamochi, G. Horiuchi, J. Saito and J. Ulański, Synth. Met., 1999, 106, 75 CrossRef CAS; (c) S. Hornchi, H. Yamochi, G. Saito, J. K. Jeszka, A. Tracz, A. Sroczyńska and J. Ulański, Mol. Cryst. Liq. Cryst., 1997, 296, 365 CrossRef PubMed; (d) J. K. Jeszka and A. Tracz, Polym. Adv. Technol., 1992, 3, 139 CrossRef CAS PubMed.
- (a) E. Laukhina, V. Tkacheva, I. Chuev, E. Yagubskii, J. Vidal-Gancedo, M. Mas-Torrent, C. Rovira, J. Veciana, S. Khasanov, R. Wojciechowski and J. Ulanski, J. Phys. Chem. B, 2001, 105, 11089 CrossRef CAS; (b) M. Wiatrowski, E. Dobruchowska, W. Maniukiewicz, U. Pietsch, J. Kowalski, Z. Szamel and J. Ulanski, Thin Solid Films, 2010, 518, 2266 CrossRef CAS PubMed; (c) X. K. Gao, W. P. Wu, Y. Q. Liu, W. F. Qiu, X. B. Sun, G. Yu and D. B. Zhu, Chem. Commun., 2006, 2750 RSC.
- R. J. Brown, A. C. Brooks, J.-P. Griffiths, B. Vital, P. Day and J. D. Wallis, Org. Biomol. Chem., 2007, 5, 3172–3182 CAS.
- C. Wang, A. S. Batsanov, M. R. Bryce and J. A. K. Howard, Synthesis, 1998, 1615 CrossRef CAS PubMed.
- G. J. Marshallsay, M. R. Bryce, G. Cooke, T. Jørgensen, J. Becher, C. D. Reynolds and S. Wood, Tetrahedron, 1993, 49, 6849 CrossRef CAS.
- N. Svenstrup and J. Becher, Synthesis, 1995, 215 CrossRef CAS PubMed.
- J. Broggi, N. Joubert, S. Dez-González, S. Berteina-Raboin, T. Zevaco, S. P. Nolan and L. A. Agrofoglio, Tetrahedron, 2009, 65, 1162 CrossRef CAS PubMed.
- (a) R. P. Schibaeva and E. B. Yagubskii, Chem. Rev., 2004, 104, 5347 CrossRef PubMed; (b) M. A. Beno, U. Geiser, K. L. Kostka, H. H. Wang, K. S. Webb, M. A. Firestone, K. D. Carlson, L. Nunez, M. H. Whangbo and J. M. Williams, Inorg. Chem., 1987, 26, 1912 CrossRef CAS.
- (a) S.-X. Liu, A. Neels, H. Stoecki-Evans, J. D. Wallis, M. Pilkington and S. Decurtins, Polyhedron, 2005, 23, 1185 CrossRef PubMed; (b) L. Martin, M. Guziak and J. D. Wallis, 2015, manuscript in preparation.
- (a) S. Wang, P. Day, J. D. Wallis, P. N. Horton and M. B. Hursthouse, Polyhedron, 2006, 25, 2583 CrossRef CAS PubMed; (b) D. A. Clemente and A. Marzotto, J. Org. Chem., 1996, 6, 941 CAS.
- (a) J. Jang, S. H. Kim, S. Nam, D. S. Chung, C. Yang, W. M. Yun, C. E. Park and J. B. Koo, Appl. Phys. Lett., 2008, 92, 143306 CrossRef PubMed; (b) G. Gu, M. G. Kane, J. E. Doty and A. H. Firester, Appl. Phys. Lett., 2005, 87, 243512 CrossRef PubMed.
- E. Laukhina, V. Tkacheva, S. Khasanov, L. Zorina, J. Gómez-Segura, A. P. D. Pino, J. Veciana, V. Laukhin and C. Rovira, ChemPhysChem, 2006, 7, 920 CrossRef CAS PubMed.
- A. Tracz, J. K. Jeska, J. Ulański, T. Pakula and J. P. Rabe, Synth. Met., 1998, 94, 17 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1052024–1052028. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra04568a |
| This journal is © The Royal Society of Chemistry 2015 |