
Spin–orbit coupling analyses of phosphorescent processes in Ir(Zppy)3 (Z = NH2, NO2 and CN)†
Abstract
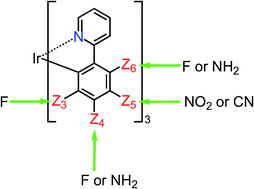
Substituent effects of NH2, NO2 and CN groups on phosphorescence in fac-tris(2-phenylpyridinato)iridium(III) [fac-Ir(ppy)3] were examined theoretically by using the multiconfiguration self-consistent field (MCSCF) method together with the SBKJC basis sets augmented by a set of polarization functions, followed by second-order configuration interaction (SOCI) and spin–orbit coupling (SOC) calculations, while time-dependent density functional theory (TD DFT) calculations provided too long wavelengths for phosphorescent peaks at the geometries optimized for triplet states even though the TD DFT predictions were qualitatively good with respect to relative spectral shifts. The strongest electron-donating substituent NH2 and the strongest electron-withdrawing substituents, NO2 and CN, were chosen for investigation of the substituent effects in the present investigation. It was found that when these electron-withdrawing substituents are introduced into the Z5 sites, the largest blue shift is obtained for the emission spectra, while the introduction of the electron-donating NH2 substituent causes a red shift of the emission spectra. This is because the Z5 site has non-negligible coefficients in the highest occupied molecular orbital (HOMO) and can interact with the π* orbitals of the substituents. This interaction makes the HOMO lower in energy. This is the reason why a large blue shift of the emission peak is obtained when one of these substituents is introduced to the Z5 sites. Based on the results of the calculation, it can be said that the best material for blue-color emission is tris(5-nitro-2-phenylpyridinato) iridium(III) [fac-Ir(5-NO2ppy)3] or tris(5-nitro-4,6-difluoro-2-phenylpyridinato)iridium(III) [fac-Ir(5-NO2-4,6-dfppy)3]. If the reactivity of the NO2 substituent in the lowest triplet state becomes troublesome in the synthesis processes and/or if it is difficult to choose host molecules for an emissive layer, tris(5-cyano-3,4,6-trifluoro-2-phenylpyridinato)iridium(III) [fac-Ir(5-CN-3,4,6-tfppy)3] would be the most appropriate for blue-color emission.
 Please wait while we load your content...
Please wait while we load your content...

