
Enhanced thermal properties in a hybrid graphene–alumina filler for epoxy composites†
Abstract
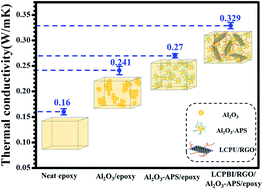
In this report, we synthesized a new kind of liquid-crystal perylenebisimide polyurethane (LCPBI). Noncovalently functionalized reduced graphene oxide (RGO) with LCPBI was prepared via π–π stacking interactions. The noncovalently functionalized graphene nanosheets (LCPBI/RGO) were used to improve the thermal properties of epoxy composites, with modified Al2O3 nanoparticles (Al2O3-APS) which were grafted with a silane coupling agent (KH-550). We demonstrated that the thermal conductivity of the epoxy composites could be improved by hybrid LCPBI/RGO and Al2O3-APS fillers. For instance, the thermal conductivity of the epoxy composite with 30 wt% Al2O3-APS and 0.3 wt% LCPBI/RGO was 0.329 W m−1 K−1, increased by 105.6% compared to that of the pure epoxy (0.16 W m−1 K−1). Meanwhile the glass transition temperature and storage modulus of epoxy composites with increasing hybrid fillers was improved, as well as the α-relaxation apparent activation energy.
 Please wait while we load your content...
Please wait while we load your content...

