
Convenient synthesis of 2,3-disubstituted quinazolin-4(3H)-ones and 2-styryl-3-substituted quinazolin-4(3H)-ones: applications towards the synthesis of drugs†
Dinesh Kumar,
Pradeep S. Jadhavar,
Manesh Nautiyal,
Himanshu Sharma,
Prahlad K. Meena,
Legesse Adane,
Sahaj Pancholia and
Asit K. Chakraborti*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Sector 67, S. A. S. Nagar 160 062, Punjab, India. E-mail: akchakraborti@niper.ac.in
First published on 19th March 2015
Abstract
Simple, convenient, and green synthetic protocols have been developed for the one pot synthesis of 2,3-disubstituted quinazolin-4(3H)-ones and 2-styryl-3-substituted quinazolin-4(3H)-ones under catalyst and solvent free conditions. The multicomponent reaction (3-MCR) involving isatoic anhydride, an amine, and orthoester afforded the 2,3-disubstituted quinazolin-4(3H)-ones in excellent yields under classical heating at 120 °C for 5 h or under microwave irradiation at 140 °C for 20–30 min. The use of ammonium acetate instead of the amine provides the 2-substituted quinazolin-4(3H)-ones. The reactions are compatible with various substituted isatoic anhydrides, aryl/heteroaryl/alkyl/cycloalkyl amines, and orthoesters. The strategies are extended to the one pot tandem condensation involving isatoic anhydride, an amine, orthoester, and aldehyde to afford highly functionalized (E)-3-aryl/heteroaryl-2-styrylquinazolin/(2-(heteroaryl)vinyl)quinazolin-4(3H)-ones. The applications of the methodologies are demonstrated through the synthesis of various drugs which act on the central nervous system such as methaqualone, mebroqualone, mecloqualone, piriquialone, and diproqualone.
Introduction
Functionalised quinazolinones are highly sought after targets for synthetic medicinal/organic chemists due to the broad spectrum of biological activities exhibited by compounds bearing this scaffold (Fig. 1).1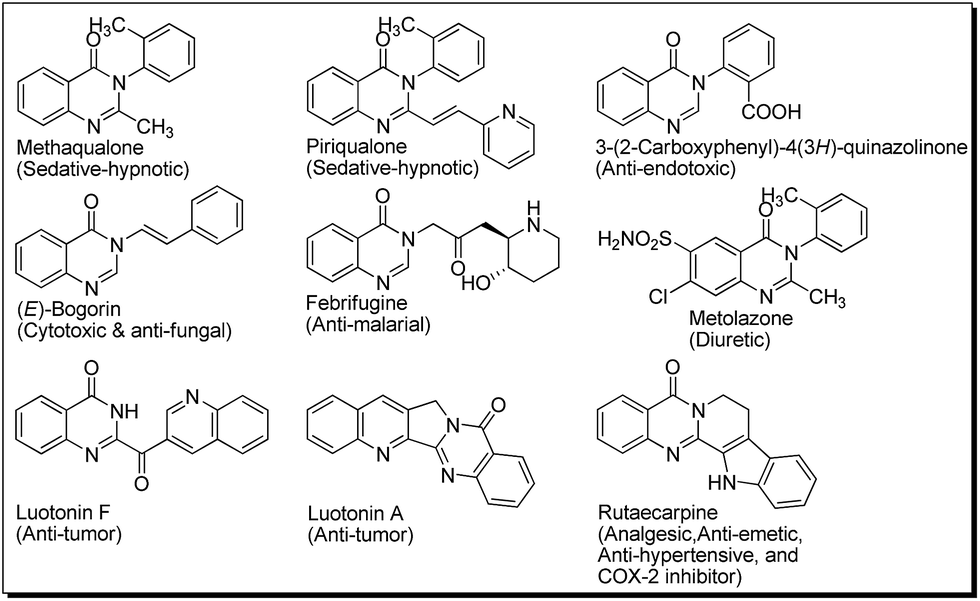 | ||
| Fig. 1 Some pharmaceutically important quinazolinones. | ||
This spurred interest to develop synthetic methodologies for this privileged pharmacophore for the generation of new therapeutic leads. The various synthetic strategies (Scheme 1) adopted for the generation of 2-methyl-3-substituted quinazolie-4(3H)-ones can be classified by the following routes: (i) reaction of N-(2-aminobenzyl) substituted amines with acetaldehyde followed by benzylic oxidation and oxidative dehydrogenation using potassium iodide-tert-butyl hydroperoxide2 (Route A), (ii) domino coupling/condensation cyclization process involving 2-halo benzamide with amidine catalyzed by CuI3 (Route B), (iii) acid catalyzed rearrangement of 4-imino-4H-3,1-benzoxazine4 (Route C), (iv) reaction of N-acetyl anthranilic acid acetic anhydride followed by the acid catalysed reaction of the intermediate formed 4H-3,1-benzoxazine with amine5 (Route D), (v) reaction of 2-nitro benzoyl chloride with amine followed by nitro reduction and its subsequent reaction with orthoester1k (Route E), (vi) reaction of anthranilic acid, amine and acetic acid6 (Route F), (vii) reaction of isatoic anhydride, amine and orthoester7 (Route G) and, (viii) reaction of 2-methyl-4H-benzo[1,3]oxazin-4-one with amine (Route H).8
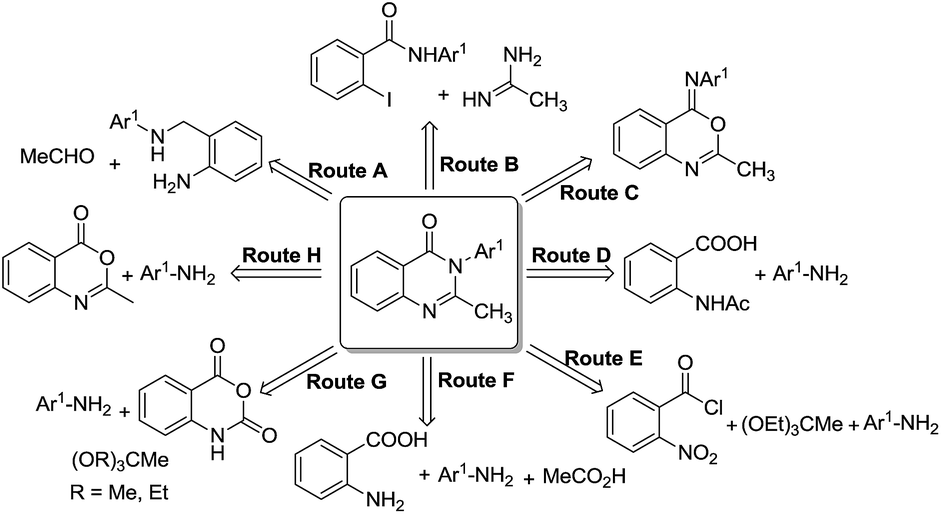 | ||
| Scheme 1 Strategies for synthesis of 2-methyl-substituted quinazolin-4(3H)-ones. | ||
On the other hand, the available methodologies for the preparation of 2-styryl quinazolin-4(3H)-ones (Scheme 2) are limited to the: (i) reaction of anthranilic acid with styryl carboxylic acid followed by amine insertion9 (Route A) and (ii) Knoevenagel condensation of 2-methyl-3-substituted quinazolin-4(3H)-one with aromatic aldehyde in the presence of LDA1b or in refluxing glacial acetic acid10 (Route B), and (iii) reaction of double lithiated 2-methyl quinazolin-4(3H)-one with aldehyde followed by TFA mediated dehydration11 (Route C).
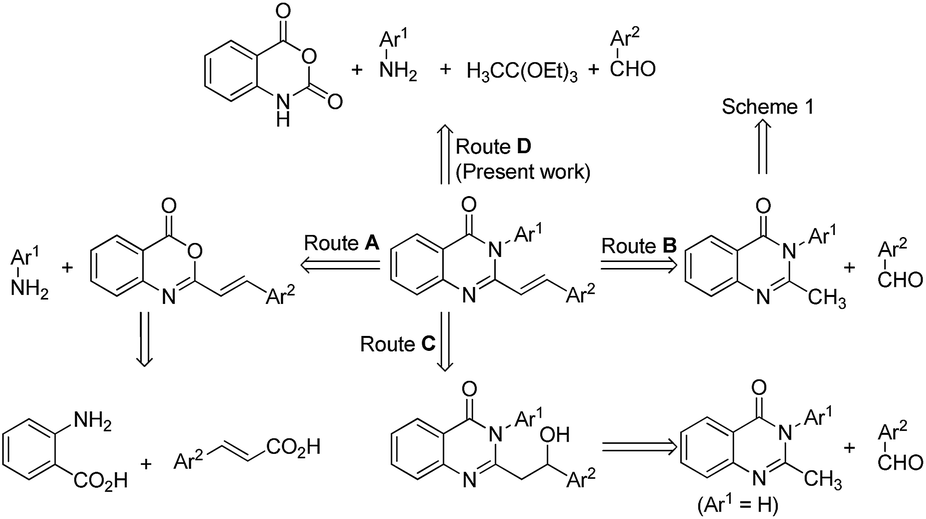 | ||
| Scheme 2 General strategies for the synthesis of 2-styryl quinazolin-4(3H)-ones. | ||
The adverse effect of the manufacturing processes of drugs and pharmaceuticals on the environment urges for sustainable development.12 The major drive towards this initiative is the replacement of the use of volatile organic solvents (VOSs) by solvent free reaction condition as VOSs are major contributors to the environmental pollution due to their abundant use (more than 85% of the total mass utilization of a chemical process) and incomplete recovery efficiency (50–80%).13 Further, the use of costly, explosive, toxic and carcinogenic chemicals (catalyst/reagents) do not qualify them under sustainability metrics as they augment the ecosystem toxicity resulting in socio-economic burden. Thus, the development of eco-friendly/green approaches to chemical synthesis is an ongoing demand. In view of the role of the N-based heterocycles as versatile pharmacophore there has been significant interest for the development of sustainable synthesis.14 In this context, solvent-free and catalyst free protocol for organic synthesis is highly desirable to comply with the triple bottomline philosophy of green chemistry,15 to meet the demand of the timely supply of the designed molecules for biological evaluation,16 and for enrichment of the medicinal chemists' tool box.17
Results and discussions
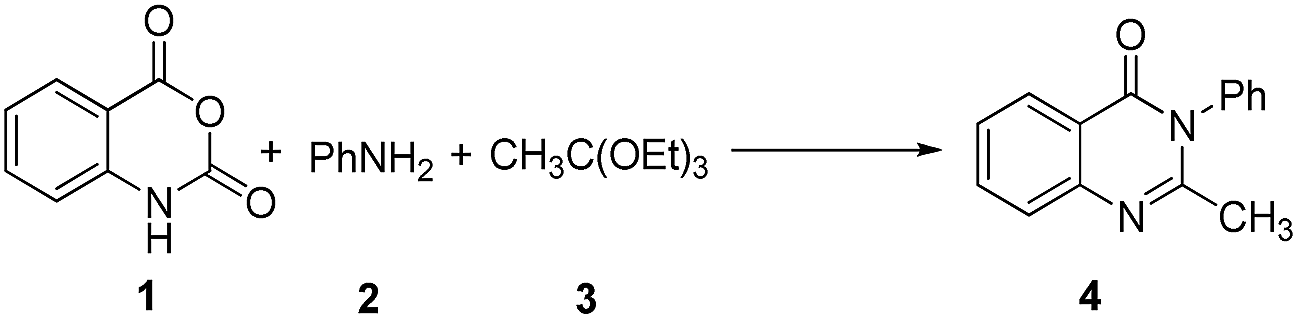
In order to develop a convenient synthesis of 2-methyl substituted quinazolinones, the model reaction involving equimolar mixture of isatoic anhydride 1, aniline 2 and triethyl orthoacetate 3 was performed under solvent and catalyst free condition at different temperature (Table 1) to form 2-methyl-3-phenylquinazolin-4(3H)-one 4. No product formation was observed at rt (25–30 °C) (entry 1, Table 1) after 6 h. However, the yield of 4 increased with the increase of the reaction temperature (entry 2–5, Table 1). The optimal result was obtained at 120 °C (entry 6, Table 1) with no further increase of yield at higher temperature (entry 7, Table 1). The best result was obtained in performing the reaction at 120 °C for 4 h (entry 9, Table 1) as lesser yield was obtained in lowering the reaction time to 2–3 h (entries 10 and 11, Table 1). To evaluate any beneficial/detrimental effect of solvents on the formation of 4, the reactions were performed in hydrocarbon, halogenated hydrocarbon, protic polar, and aprotic polar solvents under reflux but the yields were inferior compared to the solvent free condition. However, among the different solvents used, the best result was obtained in case of toluene (72%) followed by EtOH (56%).†
|
|||
|---|---|---|---|
| Entry | Temperature (°C) | Time (h) | Yieldb (%) |
| a The mixture of 1 (1 mmol), 2 (1 mmol, 1 equiv.), and 3 (1 mmol, 1 equiv.) was treated under different condition.b Isolated yield of 4. | |||
| 1 | rt (25–30) | 6 | Nil |
| 2 | 40 | 6 | Trace |
| 3 | 60 | 6 | 22 |
| 4 | 80 | 6 | 56 |
| 5 | 100 | 6 | 68 |
| 6 | 120 | 6 | 85 |
| 7 | 150 | 6 | 85 |
| 8 | 120 | 5 | 85 |
| 9 | 120 | 4 | 85 |
| 10 | 120 | 3 | 66 |
| 11 | 120 | 2 | 54 |
The requirement of longer time (4 h) led to search for an alternative mode in performing the reaction to minimise the reaction time. The microwave-assisted synthesis offers means towards green and rapid synthesis18 and has been used in this laboratory for various organic reactions including the synthesis of heterocycles.19 Therefore, the model reaction of 1, 2 and 3 was performed under microwave heating with variation of the temperature and reaction time. The best reaction condition was found to be the use of 150 W input power at 140 °C for 20 min affording 4 in 90% yield†.
Thus, two methods for solvent and catalyst-free synthesis of 2-methyl substituted quinazolinones adopting the 3-MCR strategy are developed by the treatment of isatoic anhydride, amine, and orthoacetate under: (i) conventional heating at 120 °C (oil-bath) for 4 h (Method A) and (ii) microwave heating at 140 °C for 20 min (Method B).
The versatility of these two methodologies can be demonstrated through the reaction involving varieties of isatoic anhydrides, amines, and orthoesters under conventional (Method A, Table 2) and microwave (Method B, Table 2) heating. Various aryl/heteroaryl/alkyl amines react smoothly with isatoic anhydrides and orthoesters to form the corresponding 2,3-disubstituted quinazolin-4(3H)-ones in excellent yields. However, inferior yields were obtained from amines having electron withdrawing substituents (entry 17 & 18, Table 2) compared to the amines bearing electron donating substituents (entry 10 & 11, Table 2). The reaction conditions are compatible with heterocyclic (entry 19–21, Table 2), arylalkyl (entry 22–25, Table 2), alicyclic (entry 26, Table 2), and allyl (entry 27, Table 2) amines.
|
|||||
|---|---|---|---|---|---|
| Entry | Isatoic anhydride | R | Amine/NH4OAc | Yieldb (%) | |
| Method A | Method B | ||||
| a Method A: isatoic anhydride (2.5 mmol) was treated with the amine/NH4OAc (2.5 mmol, 1 equiv.) and orthoester (2.5 mmol, 1 equiv.) under neat condition at 120 °C for the 5 h. Method B: isatoic anhydride (2.5 mmol) was treated with the amine/NH4OAc (2.5 mmol, 1 equiv.) and orthoester (2.5 mmol, 1 equiv.) under neat condition at 140 °C (150 W) for 20–30 min.b Isolated yield of the desired quinazolinone. | |||||
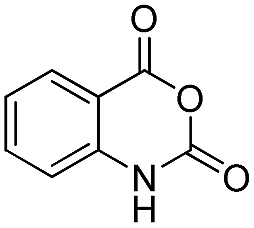 |
|||||
| 1 | H | NH4OAc | 91 | 92 | |
| 2 | Me | NH4OAc | 91 | 90 | |
| 3 | Et | NH4OAc | 90 | 91 | |
| 4 | nBu | NH4OAc | 83 | 90 | |
| 5 | Ph | NH4OAc | 78 | 86 | |
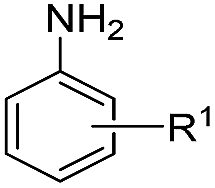 |
|||||
| 6 | H | R1 = H | 90 | 92 | |
| 7 | Me | R1 = H | 85 | 88 | |
| 8 | H | R1 = 4-Me | 90 | 90 | |
| 9 | Me | R1 = 4-Me | 86 | 87 | |
| 10 | H | R1 = 4-OMe | 92 | 91 | |
| 11 | Me | R1 = 4-OMe | 88 | 90 | |
| 12 | H | R1 = 2-Cl | 85 | 86 | |
| 13 | H | R1 = 4-Cl | 85 | 88 | |
| 14 | Me | R1 = 4-Cl | 82 | 85 | |
| 15 | Me | R1 = 4-Br | 81 | 82 | |
| 16 | Me | R1 = 4-F | 82 | 81 | |
| 17 | H | R1 = 4-CF3 | 72 | 80 | |
| 18 | Me | R1 = 4-CF3 | 70 | 79 | |
| 19 | H | 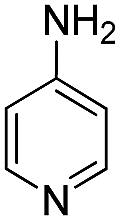 |
78 | 81 | |
| 20 | H | 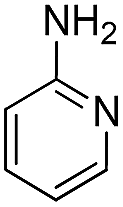 |
70 | 72 | |
| 21 | Me | 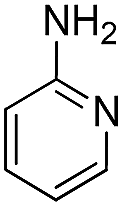 |
68 | 71 | |
| 22 | H | 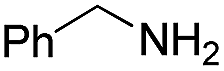 |
72 | 75 | |
| 23 | Me | 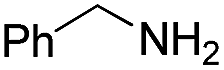 |
70 | 72 | |
| 24 | H | 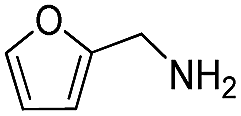 |
76 | 76 | |
| 25 | Me | 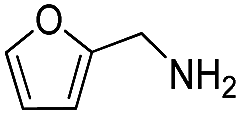 |
72 | 75 | |
| 26 | H | 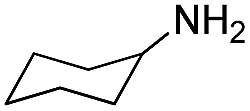 |
70 | 73 | |
| 27 | Me | 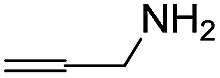 |
78 | 82 | |
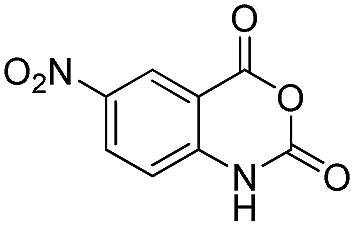 |
|||||
| 28 | Me | R1 = H | 82 | 85 | |
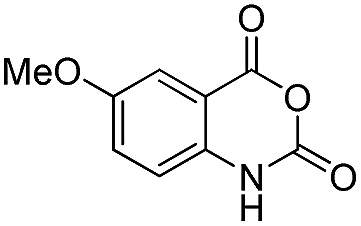 |
|||||
| 29 | Me | R1 = H | 69 | 73 | |
| 30 | Me | R1 = 4-CH3 | 67 | 70 | |
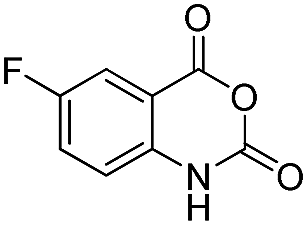 |
|||||
| 31 | Me | R1 = H | 80 | 81 | |
| 32 | Me | R1 = 4-SCH3 | 74 | 77 | |
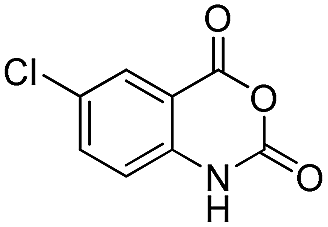 |
|||||
| 33 | Me | R1 = H | 80 | 82 | |
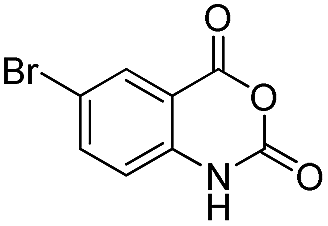 |
|||||
| 34 | Me | R1 = H | 78 | 84 | |
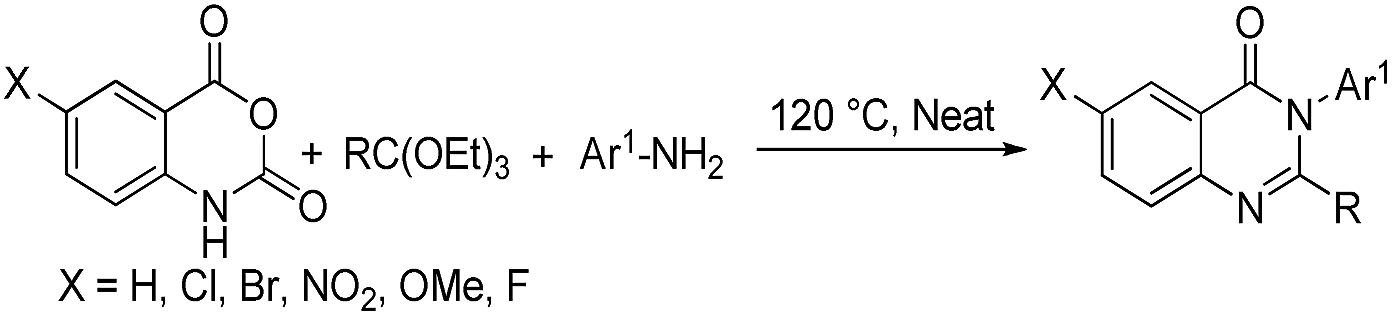
With respect to the orthoesters, the reactions were performed with commercially available orthoesters and excellent yields were obtained in each case. The reaction works well with substituted isatoic anhydrides (entries 28–34, Table 2).
The 2-methyl-3-substituted quinazolinones serve as the starting materials for the preparation of 2-styryl-3-substituted quinazolinones during the condensation with aldehydes which however requires the use of strong base such as LDA1b,11 or reflux in HOAc for prolonged period (12 h). The multicomponent reactions (MCRs)20 have been recognised as new green synthetic tools to rapidly generate the synthetic targets in convergent and atom-economical fashion. Therefore, the tandem one pot reaction (Route D, Scheme 3)21 of 1, 2, 3, and benzaldehyde 5 to form the 2-styryl-3-phenyl quinazolinone 6 was planned that would involve a 3-MCR process involving 1, 2, 3 followed by condensation of 5 with the in situ generated 4 in tandem. The only report for the preparation of 2-styryl quinazolinones following this strategy requires the use of 50 mol% of the protic ionic liquid [Hmim]TFA as the promoter.21a
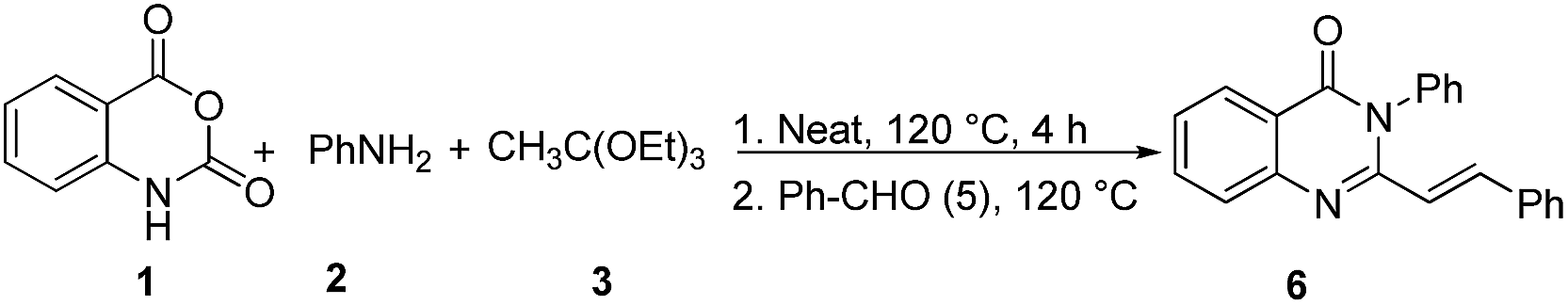 | ||
| Scheme 3 The one-pot tandem synthesis of 3-phenyl-2-styrylquinazolin-4(3H)-one. | ||
To test the feasibility, the model reaction was performed using optimized reaction conditions (conventional and microwave heating) for the condensation of 1, 2, 3, to form 4 followed by the addition of benzaldehyde 5 (1 equiv.) and stirring the reaction mixture for further 5 h. This one pot process proved to be effective under both conventional and microwave heating (entry 1, Table 3). Thus, it was realized that these two steps can be effectively carried out in one-pot to afford varieties of 2-styryl quinazolinones. The different combinations of amines and aldehydes were opted to get diversified synthesis of 2-styryl-4(3H)-quinazolinones (Table 3). The aromatic aldehydes having different functional groups as well as hetero-aromatic aldehydes worked adequately under the optimized condition giving highly functionalized 2-styryl-4(3H)-quinazolinones.
|
|||
|---|---|---|---|
| Entry | Product | Yieldb (%) | |
| Method A | Method B | ||
| a Method A: the mixture of isatoic anhydride (2.5 mmol), amine/NH4OAc (2.5 mmol) and triethyl orthoacetate (2.5 mmol) was heated under neat condition at 120 °C for 5 h followed by addition of aldehyde (2.5 mmol, 1 equiv.) and continued stirring for further 5 h. Method B: the mixture of isatoic anhydride (2.5 mmol), amine/NH4OAc (2.5 mmol) and triethyl orthoacetate (2.5 mmol) was heated under neat condition at 140 °C (150 W) for 20 min followed by addition of aldehyde (2.5 mmol, 1 equiv.) and continued for further 20–30 min.b Isolated yield of the corresponding 2-styrylquinazolinone. | |||
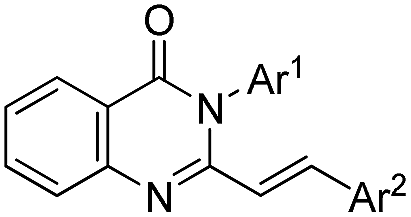 |
|||
| 1 | Ar1 = H; Ar2 = Ph | 79 | 81 |
| 2 | Ar1 = H; Ar2 = 4-OMe–C6H4 | 75 | 76 |
| 3 | Ar1 = Ph; Ar2 = Ph | 74 | 78 |
| 4 | Ar1 = Ph; Ar2 = 4-Me–C6H4 | 72 | 75 |
| 5 | Ar1 = Ph; Ar2 = 4-Br–C6H4 | 75 | 74 |
| 6 | Ar1 = Ph; Ar2 = 4-CF3–C6H4 | 78 | 80 |
| 7 | Ar1 = Ph; Ar2 = 4-OMe–C6H4 | 70 | 72 |
| 8 | Ar1 = Ph; Ar2 = 4-OCH2Ph–C6H4 | 70 | 75 |
| 9 | Ar1 = 4-Me–C6H4; Ar2 = 4-Cl–C6H4 | 75 | 76 |
| 10 | Ar1 = 4-OMe–C6H4; Ar2 = 4-OMe–C6H4 | 76 | 79 |
| 11 | Ar1 = 4-SMe–C6H4; Ar2 = 4-OMe–C6H4 | 74 | 76 |
| 12 | Ar1 = 4-SMe–C6H4; Ar2 = 4-NMe2–C6H4 | 77 | 76 |
| 13 | Ar1 = 4-SMe–C6H4; Ar2 = 4-Cl–C6H4 | 75 | 75 |
| 14 | Ar1 = 4-F–C6H4; Ar2 = 4-Cl–C6H4 | 71 | 77 |
| 15 |  |
70 | 72 |
| 16 | 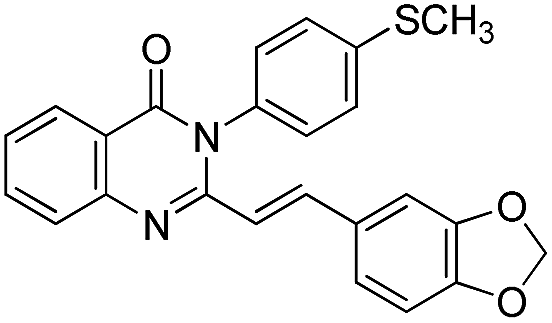 |
76 | 78 |
| 17 | 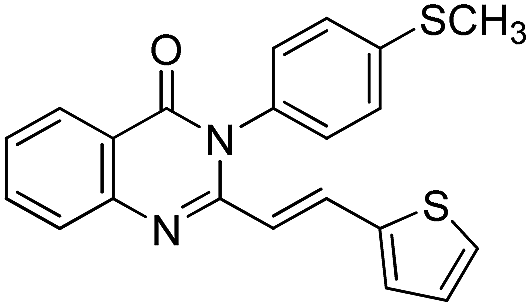 |
72 | 75 |
| 18 | 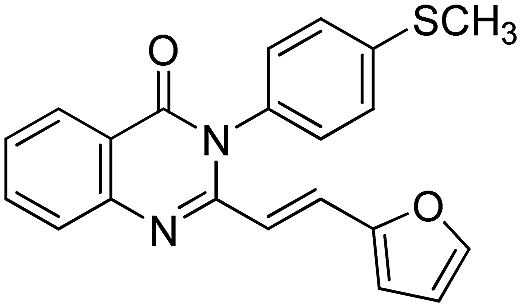 |
72 | 75 |
| 19 | 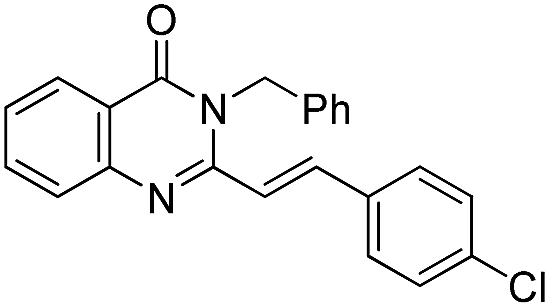 |
71 | 76 |
| 20 | 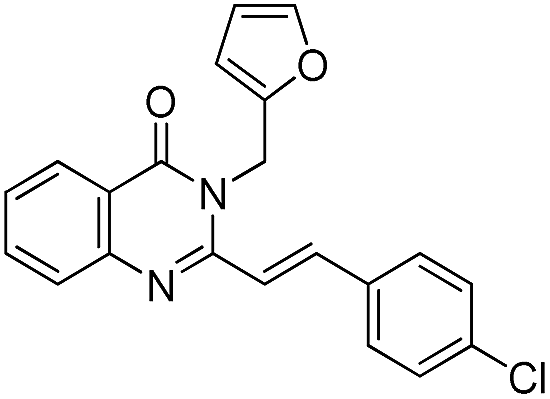 |
71 | 76 |
The ultimate utility of any synthetic protocol is judged on the basis of its use for the synthesis of target molecules. The wide applications of 4(3H)-quinazolinones and its 2-styryl derivatives as CNS depressant prompted us to test these new methodologies for the synthesis of a few CNS depressants. Thus the sedative and hypnotic drugs methaqualone 4a, mebroqualone 4b, and mecloqualone 4c were synthesized through the 3-MCR process involving isatoic anhydride, appropriate amine and triethyl orthoacetate following the Method A and Method B in very good yields (Scheme 4).
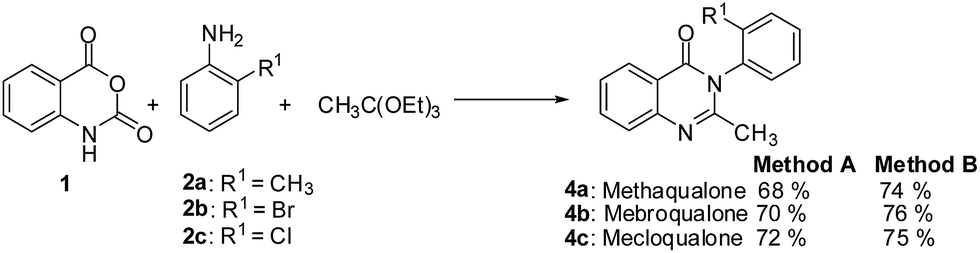 | ||
| Scheme 4 Synthesis of the sedative and hypnotic drugs 4a–c following the 3-MCR procedures. | ||
The tandem one pot procedure for the synthesis of 2-styrylquinazolinone was extended for the preparation of piriquialone 6a following the sequential addition of 1, o-toluidine 2a, 3 and pyridine-2-carboxaldehyde 5a (Scheme 5).
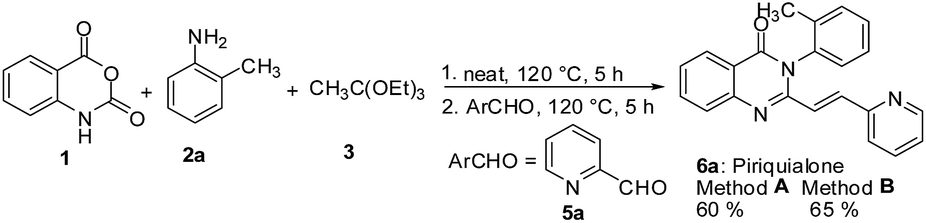 | ||
| Scheme 5 The tandem process for the synthesis of the sedative and hypnotic drug 6a. | ||
The synthesis of the sedative and hypnotic drug diproqualone 8 would involve the epoxide aminolysis22 of glycidol with the 2-methylquinazolinone 7. The requisite starting material 7 was synthesised in 90–92% yields by the newly developed 3-MCR of 1, 3, and NH4OAc. The treatment of 7 with glycidol in the presence of Zn(ClO4)2·6H2O (2 mol%)22b at room temperature under neat condition afforded 8 in 81% yield (Scheme 6).
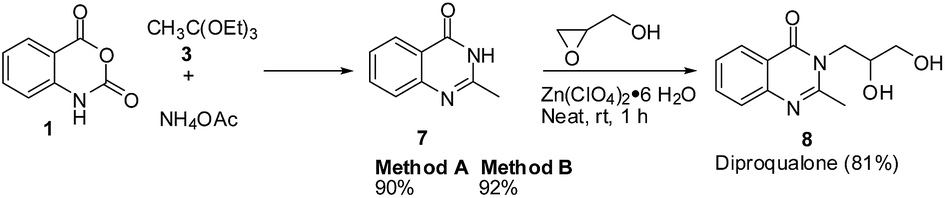 | ||
| Scheme 6 Synthesis of the sedative and hypnotic drug 8. | ||
Conclusions
The present work reports a simple and convenient synthesis of 2,3-disubstituted quinazolin-4(3H)-ones under catalyst and solvent free conditions following 3MCR strategies. The reactions are performed under classical heating at 120 °C for 5 h or under microwave dielectric heating under controlled input power, temperature, and pressure for 20–30 min. The reactions were compatible with different variations of isatoic anhydride, amine and orthoester to generate functionalized 2,3-disubstituted quinazolin-4(3H)-ones. This has been further extended for the one-pot synthesis of functionalized (E)-3-aryl/heteroaryl-2-styrylquinazolin/(2-(heteroaryl)vinyl)quinazolin-4(3H)-ones without the necessity of acid/base in the final condensation step. The catalyst and solvent free condition along with operational simplicity (recrystallisation using EtOH) offers a green process for the synthesis of quinazolin-4(3H)-ones. The synthetic utility was demonstrated through the synthesis of several CNS depressants such as methaqualone, mebroqualone, mecloqualone, piriquialone, and diproqualone in good to excellent yields.Experimental
General information
The glasswares were thoroughly washed and dried in an oven. Chemicals and all solvents were commercially available (Aldrich Chemical, Merck AG, Fluka and S-D Fine Chemicals) and used without further purification. The 1H/13C NMR spectra were recorded on a Bruker Avance 400/100 MHz NMR spectrometer in CDCl3 with residual undeuterated solvent (CDCl3: 7.26/77.0) using TMS as an internal standard. Chemical shifts (δ) are given in ppm and J values are given in Hz. 13C NMR spectra were fully decoupled and were referenced to the middle peak of the solvent CDCl3 at 77.0 ppm. Splitting pattern were designated as s, singlet; bs, broad singlet; d, doublet; dd, doublet of doublet; t, triplet; m, multiplet. Mass spectra were recorded on a Finnigan MAT-LCQ [for APCI] mass spectrometer. Infra-red (IR) spectra were recorded on Perkin Elmer FT-IR spectrometer in the range 4000–600 cm−1 either as neat samples for liquids or using KBr for preparing pellets for solid samples. Compounds were routinely checked for their purity on the silica gel GF-254 and visualized under UV at wavelength 254 nm. Melting points were measured with Gupta scientific melting point apparatus and were uncorrected. The microwave-assisted reactions were performed using CEM Discover model no 908010 equipment. Evaporation of solvents was performed at reduced pressure, using a rotary evaporator.Method A. The equimolar mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), aniline 2 (0.23 g, 2.5 mmol) and triethyl orthoacetate 3 (0.41 g, 2.5 mmol) was stirred magnetically at 120 °C (oil bath temp). After completion of the reaction (TLC, 4 h), the crude reaction mixture was recrystallized from EtOH to obtain analytically pure 4 (0.50 g, 85%) as white solid; mp = 144–145 °C (lit7a 144–146 °C); IR (KBr) νmax: 2968, 1648, 1602 cm−1; 1H NMR (400 MHz, CDCl3): δ = 8.27 (d, J = 7.5 Hz, 1H), 7.76 (d, J = 7.2 Hz 1H), 7.68 (d, J = 7.6 Hz, 1H), 7.51 (m, 4H), 7.27 (d, J = 6.0 Hz, 2H), 2.24 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 162.3, 154.2, 147.5, 137.8, 134.6, 130.0, 129.3, 128.0, 127.0, 126.8, 126.6, 120.8, 24.4; MS(APCI) m/z: 237.33 (M + H)+.
Method B. The equimolar mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), aniline 2 (0.23 g, 2.5 mmol) and triethyl orthoacetate 3 (0.41 g, 2.5 mmol) was subjected to microwave irradiation (150 W/140 °C) for 20 min. After completion of reaction, the crude reaction mixture was recrystallized from EtOH to obtain analytically pure 4 (0.52 g, 88%) as white solid.
Method A. The equimolar mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), aniline 2 (0.23 g, 2.5 mmol) and triethyl orthoacetate 3 (0.41 g, 2.5 mmol) was stirred magnetically at 120 °C (oil bath temp) for 4 h followed by addition of benzaldehyde 5 (0.26 g, 2.5 mmol) and the stirring was continued for another 4 h (TLC). The crude reaction mixture was recrystallized from EtOH to obtain analytically pure 6 (0.64 g, 79%) as white solid; mp = 196–197 °C (lit21 196 °C); IR (KBr) νmax: 3442, 1676, 1545, 1472, 1258, 1132 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 8.30 (d, J = 7.9 Hz, 1H), 7.97 (d, J = 15.5 Hz, 1H), 7.79 (d, J = 3.4 Hz, 2H) 7.53–7.61 (m, 3H), 7.47–7.50 (m, 1H), 7.28–7.34 (m, 7H), 6.39 (d, J = 15.5 Hz, 1H); MS (APCI) m/z: 325.21 (M + H)+.
Method B: (entry 3, Table 3). The equimolar mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), aniline 2 (0.23 g, 2.5 mmol) and triethyl orthoacetate 3 (0.41 g, 2.5 mmol) was subjected to microwave irradiation (150 W/140 °C) for 20 min followed by addition of benzaldehyde 5 (0.26 g, 2.5 mmol) and continued the reaction for another 20 min. The crude reaction mixture was recrystallized using EtOH to obtain analytically pure 6 (0.66 g, 81%) as white solid.
2-Methyl-3-o-tolylquinazolin-4(3H)-one 4a. The equimolar mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), triethyl orthoacetate 3 (0.41 g, 2.5 mmol) and 2-methyl aniline (0.21 g, 2.5 mmol) was stirred magnetically at 120 °C (oil bath temp). After completion of reaction (TLC, 4 h), the crude reaction mixture was recrystallized from EtOH to obtain analytically pure 4a (0.43 g, 68%) as white solid; mp = 118–119 °C (lit1b 114–115 °C); IR (KBr) νmax = 3008, 1683, 1600, 1471, 1274 cm −1; 1H NMR (400 MHz, CDCl3) δ: 8.29 (d, J = 7.9 Hz, 1H), 7.78 (t, J = 7.6 Hz, 1H), 7.69 (d, J = 8.2 Hz, 1H), 7.47 (t, J = 7.5 Hz, 1H), 7.41–7.36 (m, 3H), 7.16 (d, J = 7.3 Hz, 1H), 2.19 (s, 3H), 2.13 (s, 3H); MS(APCI) m/z 251.41 (M + H)+.
Synthesis of 3-(2-bromophenyl)-2-methylquinazolin-4(3H)-one 4b. The equimolar mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), triethyl orthoacetate 3 (0.41 g, 2.5 mmol) and 2-bromo aniline (0.28 g, 2.5 mmol) was stirred magnetically at 120 °C (oil bath temp). After completion of reaction (TLC, 4 h), the crude reaction mixture was recrystallized from EtOH to obtain analytically pure 4b (0.54 g, 70%) as white solid; mp = 143–145 °C (lit1b 143–144 °C); IR (KBr) νmax = 2923, 1687, 1607, 1471, 1281 cm −1; 1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J = 7.9 Hz, 1H), 7.79–7.76 (m, 2H), 7.70 (d, J = 7.5 Hz, 1H), 7.51–7.47 (m, 2H), 7.50–7.46 (m, 3H), 7.40–7.36 (m, 2H), 2.22 (s, 3H); MS(APCI) m/z 271.24 (M + H)+.
Synthesis of 3-(2-chlorophenyl)-2-methylquinazolin-4(3H)-one 4c. The equimolar mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), triethyl orthoacetate 3 (0.41 g, 2.5 mmol) and 2-chloro aniline (0.32 g, 2.5 mmol) was stirred magnetically at 120 °C (oil bath temp). After completion of reaction (TLC, 4 h), the crude reaction mixture was recrystallized from EtOH to obtain analytically pure 4c (0.48 g, 72%) as brown solid; mp = 126–128 °C (lit1b 126–127 °C); IR (KBr) νmax = 2925, 1688, 1608, 1471, 1281 cm −1; 1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J = 6.8 Hz, 1H), 7.78 (t, J = 8.2 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.62–7.60 (m, 1H), 7.50–7.46 (m, 3H), 7.36–7.33 (m, 1H), 2.23 (s, 3H); MS(APCI) m/z 271.24 (M + H)+.
Synthesis of (E)-2-(2-(pyridin-2-yl)vinyl)-3-(o-tolyl)quinazolin-4(3H)-one (piriquialone) 6a. The equimolar mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), triethyl orthoacetate 3 (0.41 g, 2.5 mmol) and NH4OAc (0.23 g, 3 mmol) was stirred magnetically at 120 °C (oil bath temp) for 5 h followed by addition of pyridine-2-carboxaldehyde 5a (0.27 g, 2.5 mmol) and the stirring was continued for another 4 h (TLC). The crude reaction mixture was recrystallized from EtOH to obtain analytically pure 6a (0.50 g, 60%) as white solid; mp = 195–196 °C (lit21 195 °C); IR (KBr) νmax = 3326, 1645, 1525, 1126, 1023 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 8.50 (d, J = 3.9 Hz, 1H), 8.31–8.34 (m, 1H), 8.00 (d, J = 15.0 Hz, 1H), 7.80–7.82 (m, 2H), 7.61–7.63 (m, 1H), 7.48–7.52 (m, 1H), 7.40–7.46 (m, 3H), 7.30 (d, J = 7.8 Hz, 1H), 7.22 (d, J = 7.6 Hz, 1H), 7.14–7.18 (m, 1H), 6.92 (d, J = 15.0 Hz, 1H); MS (APCI) m/z: 340.42 (M + H)+.
Step-1 synthesis of 2-methylquinazolin-4(1H)-one 7. The mixture of isatoic anhydride 1 (0.41 g, 2.5 mmol), triethyl orthoacetate 3 (0.41 g, 2.5 mmol) and NH4OAc (0.23 g, 3 mmol, 1.2 equiv.) was stirred magnetically at 120 °C (oil bath temp). After completion of the reaction (TLC, 4 h), the crude reaction mixture was recrystallized from EtOH to obtain analytically pure 2-methylquinazolin-4(3H)-one 7 (0.36 g, 90%) as white solid; mp = 230–232 °C (lit7c 229–230 °C); IR (KBr) νmax: 1675, 1462, 1258, 750 cm−1; 1H NMR (400 MHz, CDCl3): δ = 12.07 (s, 1H), 8.29 (d, J = 7.9 Hz, 1H), 7.77 (t, J = 8.0 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.47 (t, J = 7.1 Hz, 1H), 2.60 (s, 3H); MS(APCI) m/z: 161.28 (M + H)+.
Step-2 ring opening of glycidol with 2-methylquinazolin-4(1H)-one. To a magnetically stirred mixture of 2-methylquinazolin-4(3H)-one 7 (0.16 g, 1 mmol) and oxiran-2-ylmethanol (0.07 g, 1 mmol) was added Zn(ClO4)2·6H2O (2 mol%) under neat condition at rt and the stirring was continued for 30 min. The crude reaction mixture was purified by flash column chromatography to obtain analytically pure 8 (0.18 g, 78%) as white solid; mp = 142–145 °C (lit23 142–145 °C); IR (KBr) νmax: 3132, 1632, 1613, 1543, 1390 cm−1; 1H NMR (400 MHz, CDC): δ = 8.10 (dd, J = 8.0 Hz & 1.2 Hz, 1H),7.76–7.80 (m, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.45–7.49 (m, 1H), 5.08 (d, J = 4.6 Hz, 1H), 4.79 (t, J = 5.7 Hz, 1H), 4.27–4.30 (m, 1H), 3.82–3.87 (m, 2H), 2.65 (s, 3H); MS(APCI) m/z: 235.32 (M + H)+.
Acknowledgements
DK thanks CSIR, New Delhi for award of Research Associateship. LA thanks FICCI, New Delhi for award of C V Raman Fellowship 2012 for African researchers.Notes and references
- CNS depressants: (a) C. M. Gupta, A. P. Bhaduri and N. M. Khanna, J. Med. Chem., 1968, 11, 392 CrossRef CAS; (b) J. F. Wolfe, T. L. Rathman, M. C. Sleevi, J. A. Campbell and T. D. Greenwood, J. Med. Chem., 1990, 33, 161 CrossRef CASAnalgesic: (c) S. Rádl, P. Hezky, J. Proška and I. Krejcí, Arch. Pharm., 2000, 333, 381 CrossRefAnti-bacterial: (d) P.-P. Kung, M. D. Casper, K. L. Cook, L. W. Lingardo, L. M. Risen, T. A. Vickers, R. Ranken, L. B. Blyn, J. R. Wyatt, P. Dan Cook and D. J. Ecker, J. Med. Chem., 1999, 42, 4705 CrossRef CAS PubMedAnti-diabetic: (e) M. S. Malamas and J. Millen, J. Med. Chem., 1991, 34, 1492 CrossRef CASAnti-inflammatory: (f) M. D. F. Pereira, R. Chevrot, E. Rosenfeld, V. Thiery and T. Besson, J. Enzyme Inhib. Med. Chem., 2007, 22, 577 CrossRef CAS PubMed; (g) Q. Chao, L. Deng, H. Shih, L. M. Leoni, D. Genini, D. A. Carson and H. B. Cottam, J. Med. Chem., 1999, 42, 3860 CrossRef CAS PubMedAnti-tumor: (h) D.-J. Baek, Y.-K. Park, H. I. Heo, M. Lee, Z. Yang and M. Choi, Bioorg. Med. Chem. Lett., 1998, 8, 3287 CrossRef CAS; (i) S. E. Webber, T. M. Bleckman, J. Attard, J. G. Deal, V. Kathardekar, K. M. Welsh, S. Webber, C. A. Janson and D. A. Matthews, J. Med. Chem., 1993, 36, 733 CrossRef CAS; (j) J. B. Jiang, D. P. Hesson, B. A. Dusak, D. L. Dexter, G. J. Kang and E. Hamel, J. Med. Chem., 1990, 33, 1721 CrossRef CASAnti-allergic: (k) N. P. Peet, L. E. Baugh, S. Sunder, J. E. Lewis, E. H. Matthews, E. L. Olberding and D. N. Shah, J. Med. Chem., 1986, 29, 2403 CrossRef CASAnti-hypertensive: (l) M. A. Hussain, A. T. Chiu, W. A. Price, P. B. Timmermans and E. Shefter, Pharm. Res., 1988, 5, 242 CrossRef CASAnti-malarial: (m) Y. Takaya, H. Tasaka, T. Chiba, K. Uwai, M.-a. Tanitsu, H.-S. Kim, Y. Wataya, M. Miura, M. Takeshita and Y. Oshima, J. Med. Chem., 1999, 42, 3163 CrossRef CAS PubMedNon-nucleoside reverse transcriptase inhibitor: (n) J. W. Corbett, S. S. Ko, J. D. Rodgers, L. A. Gearhart, N. A. Magnus, L. T. Bacheler, S. Diamond, S. Jeffrey, R. M. Klabe, B. C. Cordova, S. Garber, K. Logue, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, J. Med. Chem., 2000, 43, 2019 CrossRef CAS PubMedPhosphorylation inhibitor: (o) B. D. Palmer, S. Trumpp-Kallmeyer, D. W. Fry, J. M. Nelson, H. D. H. Showalter and W. A. Denny, J. Med. Chem., 1997, 40, 1519 CrossRef CAS PubMed; (p) K. Matsuno, M. Ichimura, T. Nakajima, K. Tahara, S. I. Fujiwara, H. Kase, J. Ushiki, N. A. Giese, A. Pandey, R. M. Scarborough, N. A. Lokker, J.-C. Yu, J. Irie, E. Tsukuda, S.-I. Ide, S. Oda and Y. Nomoto, J. Med. Chem., 2002, 45, 3057 CrossRef CAS PubMed.
- R. A. Kumar, C. U. Maheswari, S. Ghantasala, C. Jyothi and K. R. Reddy, Adv. Synth. Catal., 2011, 353, 401 CrossRef CAS.
- J. Zhoua, L. Fua, M. Lva, J. Liu, D. Peia and K. Ding, Synthesis, 2008, 3974 Search PubMed.
- R. Mazurkiewicz, Monatsh. Chem., 1989, 120, 973 CrossRef CAS.
- S. A. Afsah, J. Ahmad, R. Purbey and A. Kumar, Asian J. Chem., 2003, 15, 552 CAS.
- O. B. Pawar, F. R. Chavan, S. S. Sakate and N. D. Shinde, Chin. J. Chem., 2010, 28, 69 CrossRef CAS.
- (a) A. A. Mohammadi and S. S. S. Hossini, Chin. J. Chem., 2011, 29, 1982 CrossRef CAS; (b) M. Bakavoli, O. Sabzevari and M. Rahimizadeh, Chin. Chem. Lett., 2007, 18, 533 CrossRef CAS PubMed; (c) P. Salehi, M. Dabiri, M. A. Zolfigol and M. Baghbanzadeh, Tetrahedron Lett., 2005, 46, 7051 CrossRef CAS PubMed; (d) H. Hazarkhani and B. Karimi, Tetrahedron, 2003, 59, 4757 CrossRef CAS.
- I. K. Kostakis, A. Elomri, E. Seguin, M. Iannelli and T. Besson, Tetrahedron Lett., 2007, 48, 6609 CrossRef CAS PubMed.
- J.-F. Liu, M. Kaselj, Y. Isome, P. Ye, K. Sargent, K. Sprague, D. Cherrak, C. J. Wilson, Y. Si, D. Yohannes and S.-C. Ng, J. Comb. Chem., 2006, 8, 7 CrossRef CAS PubMed.
- D. Raffa, M. C. Edler, G. Daidone, B. Maggio, M. Merickech, S. Plescia, D. Schillaci, R. Bai and E. Hamel, Eur. J. Med. Chem., 2004, 39, 299 CrossRef CAS PubMed.
- I. Philipova, G. K. Dobrikov, K. Krumova and J. Kaneti, J. Heterocycl. Chem., 2006, 43, 1057 CrossRef CAS.
- M. Poliakoff and P. Licence, Nature, 2007, 450, 810 CrossRef CAS PubMed.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS.
- (a) N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trindade, Chem. Rev., 2009, 109, 2703 CrossRef CAS PubMed; (b) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, L. Buriol and P. Machado, Chem. Rev., 2009, 109, 4140 CrossRef CAS PubMed.
- P. Tundo, P. Anastas, D. S. Black, J. Breen, T. Collins, S. Memoli, J. Miyamoto, M. Polyakoff and W. Tumas, Pure Appl. Chem., 2000, 72, 1207 CAS.
- J. Potosky, Drug Discovery Today, 2005, 10(17), 115 CrossRef.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- B. A. Roberts and C. R. Strauss, Acc. Chem. Res., 2005, 38, 653 CrossRef CAS PubMed.
- (a) H. F. Motiwala, R. Kumar and A. K. Chakraborti, Aust. J. Chem., 2007, 60, 369 CrossRef CAS; (b) R. Kumar, C. Selvam, G. Kaur and A. K. Chakraborti, Synlett, 2005, 1401 CAS; (c) A. K. Chakraborti, C. Selvam, G. Kaur and S. Bhagat, Synlett, 2004, 851 CrossRef CAS PubMed; (d) A. K. Chakraborti and G. Kaur, Tetrahedron, 1999, 55, 13265 CrossRef CAS.
- (a) D. Kumar, D. N. Kommi, A. R. Patel and A. K. Chakraborti, Green Chem., 2012, 14, 2038 RSC; (b) S. Raha Roy, P. S. Jadhavar, K. Seth, K. K. Sharma and A. K. Chakraborti, Synthesis, 2011, 2261 Search PubMedFor reviews please see: (c) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (d) J. Zhu, H. Bienaymé, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed; (e) P. Wipf, C. R. J. Stephenson and K. Okumura, J. Am. Chem. Soc., 2003, 125, 14694 CrossRef CAS PubMed.
- (a) M. Dabiri, M. Baghbanzadeh and A. S. Delbari, J. Comb. Chem., 2008, 10, 700 CrossRef CAS PubMed; (b) M. Baghbanzadeh, M. Molnar, M. Damm, C. Reidlinger, M. Dabiri and C. O. Kappe, J. Comb. Chem., 2009, 11, 676 CrossRef CAS PubMed; (c) J. Jampilek, R. Musiol, J. Finster, M. Pesko, J. Carroll, K. Kralova, M. Vejsova, J. O'Mahony, A. Coffey, J. Dohnal and J. Polanski, Molecules, 2009, 14, 4246 CrossRef CAS PubMed; (d) A. Mrozek-Wilczkiewicz, D. S. Kalinowski, R. Musiol, J. Finster, A. Szurko, K. Serafin, M. Knas, S. K. Kamalapuram, Z. Kovacevic, J. Jampilek, A. Ratuszna, J. Rzeszowska-Wolny, D. R. Richardson and J. Polanski, Bioorg. Med. Chem., 2010, 18, 2664 CrossRef CAS PubMed.
- (a) B. Pujala, S. Rana and A. K. Chakraborti, J. Org. Chem., 2011, 76, 8768 CrossRef CAS PubMed; (b) S. B. Pujala and A. K. Chakraborti, J. Org. Chem., 2007, 72, 3713 CrossRef PubMed; (c) A. K. Chakraborti, A. Kondaskar and S. Rudrawar, Tetrahedron, 2004, 60, 9085 CrossRef CAS PubMed; (d) A. K. Chakraborti, S. Rudrawar and A. Kondaskar, Eur. J. Org. Chem., 2004, 3597 CrossRef CAS; (e) A. K. Chakraborti, S. Rudrawar and A. Kondaskar, Org. Biomol. Chem., 2004, 2, 1277 RSC; (f) A. K. Chakraborti and A. Kondaskar, Tetrahedron Lett., 2003, 44, 8315 CrossRef CAS PubMed; (g) For review: C. Schneider, Synthesis, 2006, 3919 CrossRef CAS.
- O. I. El-Sabbagh, S. M. Ibrahim, M. M. Baraka and H. Kothayer, Arch. Pharm., 2010, 243, 274 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Typical experimental procedure, spectral data of all compounds, scanned spectra of unknown compounds. See DOI: 10.1039/c5ra03888j |
| This journal is © The Royal Society of Chemistry 2015 |