
Model peptides containing the 3-sulfanyl-norbornene amino acid, a conformationally constrained cysteine analogue effective inducer of 310-helix secondary structures†
Abstract
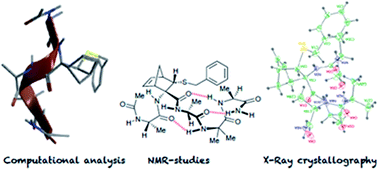
The properties of the constrained tetrasubstituted 3-sulfanylnorbornene amino acid (NRB), when inserted in Ala-Aib model peptides, were extensively studied. The conformational behaviour of these models was evaluated by theoretical calculations, spectroscopic analyses and by X-ray crystallography. Taken together, our data confirm that both (R,R,R,S)- and (S,S,S,R)-NRB enantiomers possess a strong helicogenic effect when inserted in short Ala-Aib sequences, suggesting that the rigid norbornane core has a positive effect on the ability to stabilize helical secondary structures. This information will be essential for future applications in the rational design of conformationally stable peptides targeted on protein–protein interaction (PPI) surfaces.
 Please wait while we load your content...
Please wait while we load your content...

