DOI:
10.1039/C5RA03528G
(Paper)
RSC Adv., 2015,
5, 48071-48078
MFA zeotype catalyst: a greener approach for the synthesis of INH azomethine scaffolds†
Received
26th February 2015
, Accepted 1st May 2015
First published on 1st May 2015
Abstract
Herein, we are reporting the green and efficient synthesis of some pharmacologically important azomethine derivatives of isoniazide (INH) using Modified Fly Ash (MFA) as an excellent zeotic solid acid catalyst. The catalyst, by virtue of its terminal hydroxyl groups, forms hydrogen bonds with carbonyl compounds, which activates these reactants for condensation. The MFA was assessed for various aspects like crystallinity, porosity, elemental composition, linkages and also for its stability, which were confirmed with the help of some physical spectral analyses like XRD, BET, EDS, FTIR and TGA. An effective MFA synthesis was achieved by the calcination of an aqueous mixture of fly ash with ferric chloride, successfully incorporating the iron to generate a slightly acidic crystalline zeotic material, which served as an energy efficient catalyst by allowing access to the reaction at room temperature.
Introduction
Nowadays, owing to the environmental restrictions on emissions covered in several legislations throughout the world, non-polluting and atom-efficient catalytic technologies are attracting much attention.1 The use of acid catalysts is very common in chemical and refinery industries,2 and technologies employing highly corrosive, hazardous and polluting liquid acids are being replaced by solid acids,3 for instance, acid treated clays, zeolites,4 zeotypes,5 ion-exchange resins6 and metal oxides.7
Fly ash is the solid waste residue produced from coal, oil and biomass combustion and comprises of the fine particles that rise up with the flue gases.8 It is estimated that efficient disposal of fly ash is a worldwide issue because of the huge amount produced and its harmful effects on the environment.9 Nowadays, the synthesis of zeolites with the use of fly ash is attracting lots of attention due to its peculiar properties in catalysing many reactions.10 Fly ash acts as a rich source of silica in the synthesis of zeolites, which easily combines with transition metals on hydrothermal or calcination treatment forming molecules with highly branched zeotype geometries.11
Isonicotinic acid hydrazide or isonicotinylhydrazide, commonly known as isoniazide (INH),12 is a bacteriostatic/bactericidal agent used to treat infections caused by Mycobacterium tuberculosis.13 It is a first line tubercular prodrug that is activated to isonicotinic acid on the surface of M. tuberculosis by the katG enzyme and is only active against susceptible bacteria during bacterial cell division.14 INH can form Schiff bases with carbonyl compounds and their metal chelates exhibit anticancer activity.15 The azomethine derivatives of INH, derived from its condensation with 3,4-dimethoxy benzaldehyde and 3,4,5-trimethoxy benzaldehyde, are known to show antimicrobial activity,16 while those derived from 3-methoxy-4-hydroxy benzaldehyde and 4-oxypropane benzaldehyde are known as the most potent antioxidants with significant hydrogen peroxide scavenging ability.6
The present study throws light on the development of a new catalyst by the modification of fly ash, the waste generated in thermal power plants. The catalyst is found to be highly energy efficient in the synthesis of azomethine derivatives of INH. Reports up to now have utilized acetic acid,17 and hydrochloric acid as the catalyst at reflux and there are also some reports that are without a catalyst but that reflux for at least for 2–3 hours.18 Hence, looking into the wider scope, the development of new molecular entities with special reference to INH azomethine scaffolds using MFA at RT is reported in the present study.
Experimental
Materials
All the materials (like ferric chloride, urea, INH and the aryl carbonyl compounds) were purchased from SD Fine Chemicals Pvt. Ltd, Mumbai (India). The fly ash was collected from a thermal power plant in the nearby area; Deep Nagar, Tal- Bhusawal, Dist – Jalgaon – 425![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 001 (MS), India. The fly ash was sampled in equal volumes from four areas of the plant and mixed thoroughly to make a homogeneous mass.
001 (MS), India. The fly ash was sampled in equal volumes from four areas of the plant and mixed thoroughly to make a homogeneous mass.
Methods
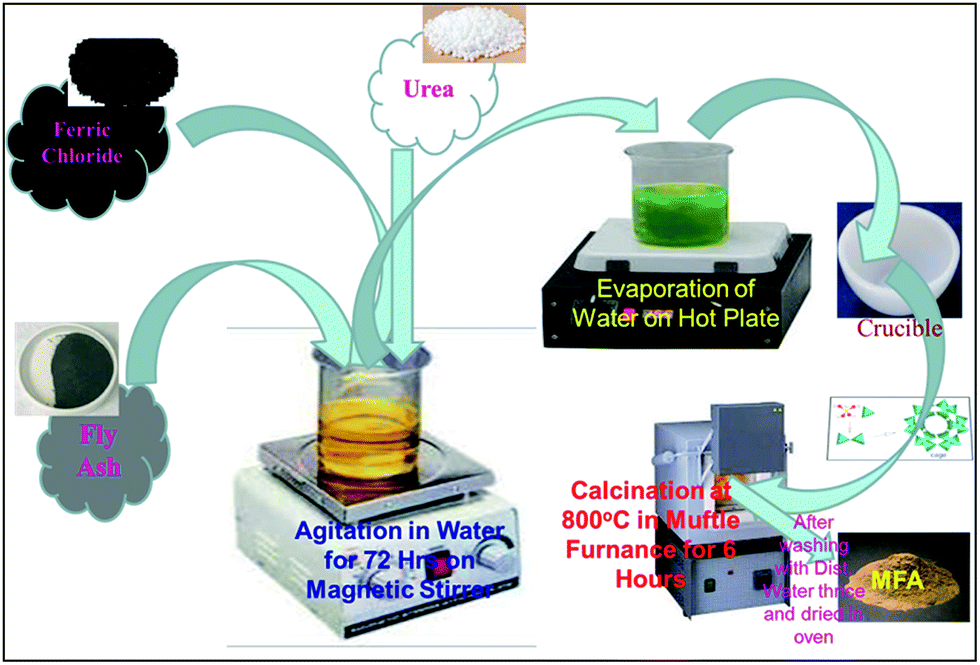
Synthesis of the MFA zeotype catalyst
 |
| | Fig. 1 Schematic diagram of MFA synthesis. | |
Reaction scheme
INH (10 mmol), aromatic aldehydes or ketones (10 mmol) (2a–h) and a catalytic amount of MFA (10% w/w of reaction mass) were taken up in EtOH and mixed thoroughly (Scheme 1). The condensation reaction at RT yielded the N-(substituted benzylidene) isonicotinohydrazide derivatives in 10–30 min (3a–h). In the synthesis, firstly a measured quantity of INH was added to a round bottom flask, followed by the addition of a measured quantity of aldehyde or ketone. Then, the mixture was mixed thoroughly on stirring and the addition of MFA was executed. The reaction was continuously monitored using a TLC technique to observe the formation of product. After that, the reaction mass was filtered through Whatman filter paper and washed with water. The obtained product was then recrystallized from pure ethanol; the separated MFA residue was first removed using filter paper and the pure product appeared in the filtrate on cooling.
 |
| | Scheme 1 Synthesis of (E)-N′-(substituted-benzylidene) isonicotinohydrazide derivatives. | |
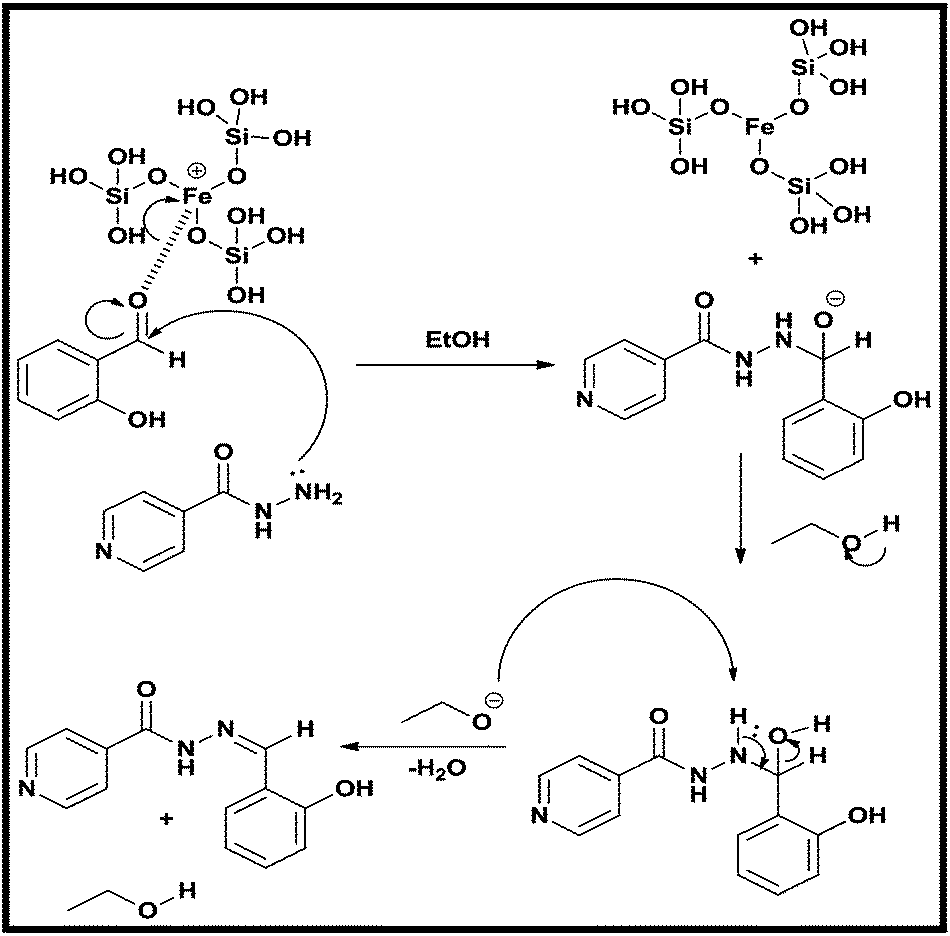
Proposed mechanism
The mechanism proposed reveals the catalytic role of MFA in the synthesis of azomethine derivatives of INH. The core functional group responsible for its catalytic behaviour is ferrous silicate, having the Fe–O–Si linkages present on the surface and in the pore voids of MFA polarizes the aldehydes or ketones and increases the electron deficiency at their carbonyl carbon centre. The lone pair of the amine nitrogen present in INH easily attacks a sufficiently electron deficient carbonyl carbon, followed by the abstraction of an acidic proton from the solvent and further dehydration leading to formation of the azomethine derivatives of INH.
Optimization of the efficient amount of MFA required for an excellent yield of 3a
The reaction was tested with different amounts of catalyst to establish the optimal amount of catalyst required for the best results in terms of time and yield in the synthesis of (E)-N′-benzylideneisonicotinohydrazide (3a). The study (Chart 1) shows that a low amount of catalyst lead to a long reaction time, while higher amounts resulted in a decrease in the yield of the reaction. The optimal amount was found to be 10% w/w of the reaction mass.
 |
| | Chart 1 | |
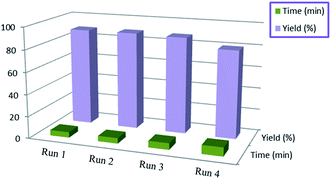
Catalyst reusability study (3a)
The catalyst was subjected to a study on its reusability in the synthesis of (E)-N′-benzylideneisonicotinohydrazide (3a) (Chart 2). The MFA recovered by filtration from the previous step was firstly washed with hot EtOH and water. Then, the purified MFA was subjected to activation at 120 °C in an oven for 4 hours. The activated MFA was assessed for its catalytic efficiency again in the next set of reactions. The study revealed that the MFA could be used efficiently up to 3 times after its recovery from previous syntheses.
 |
| | Chart 2 | |
Results and discussions
Characterization of the MFA
IR analysis. The MFA has terminal –OH groups as estimated by IR spectroscopy (Fig. 2), with peaks at 3500 cm−1 to 3300 cm−1. The peaks at 1153 cm−1 and 1103 cm−1 are attributed to Si–O linkages while the peak at 894 cm−1 is evidence of the presence of an Fe–O bond. The bonds at 800 cm−1, 790 cm−1 and 594 cm−1 show the presence of Fe–O–Si linkages.
 |
| | Fig. 2 FTIR of MFA. | |
XRD analysis. As already concluded from the IR analysis (Fig. 2) and EDXS analysis, the incorporation of iron metal as iron silicate was executed successfully. No modification of the XRD pattern (Fig. 3) by the replacement of aluminium or other trace impurities with iron was evident, so the geometry of the crystal lattice was conserved. The X-ray diffractogram was produced on a Bruker D8 ADVANCE instrument with Cu-Kα radiation (λ = 0.514 nm). The X-ray diffraction pattern was recorded over 0 to 80° (2θ degree).
 |
| | Fig. 3 Powder XRD analysis. | |
FESEM analysis. FESEM images (Fig. 4 and 5) were taken on a Hitachi S4800 instrument after gold plating was applied with the help of a Hitachi Ion Sputter E1010 instrument. The FESEM analyses show that the MFA has a spherical geometry. The particles have diameters in the range of 1 μm to 10 μm, which is in accordance with a spherical dimension.
 |
| | Fig. 4 FESEM image of MFA. | |
 |
| | Fig. 5 FESEM image of MFA. | |
EDXS analysis. The EDXS spectra recorded on a Bruker X Flash Detector show the successful incorporation of iron into the fly ash, collected from the thermal power plant, to form mesoporous ferrous silicates. The composition contains 8% iron and 17% aluminium, while silica and oxygen are present in 25% and 50% respectively.
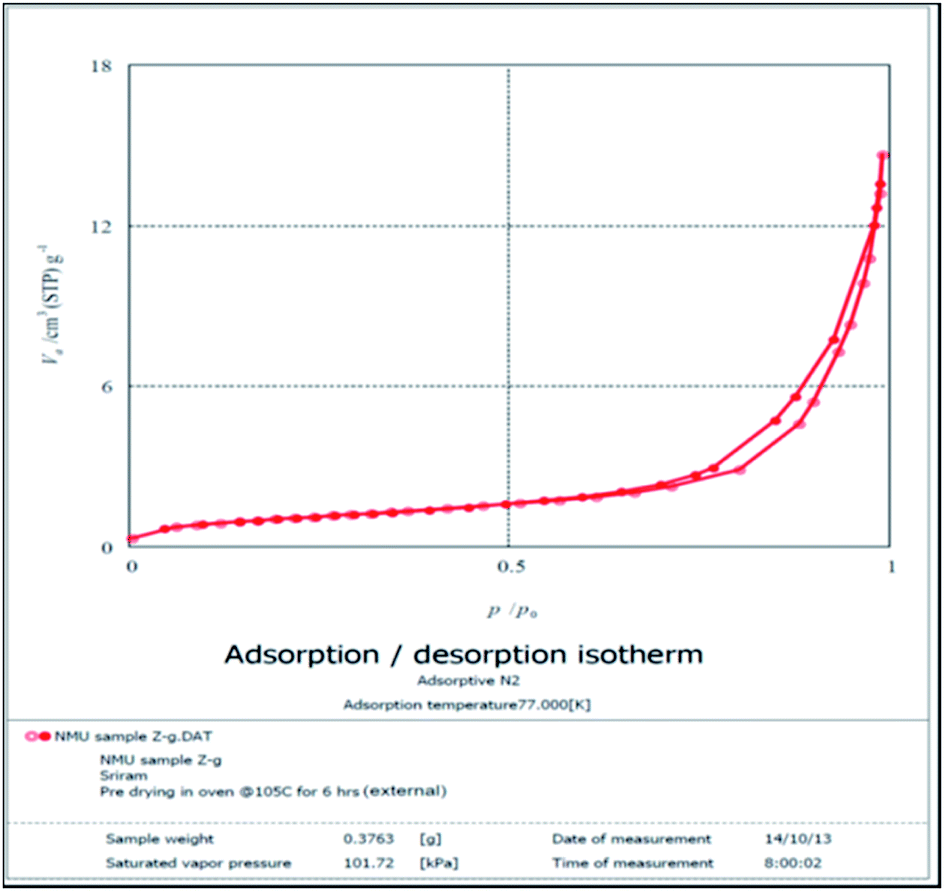
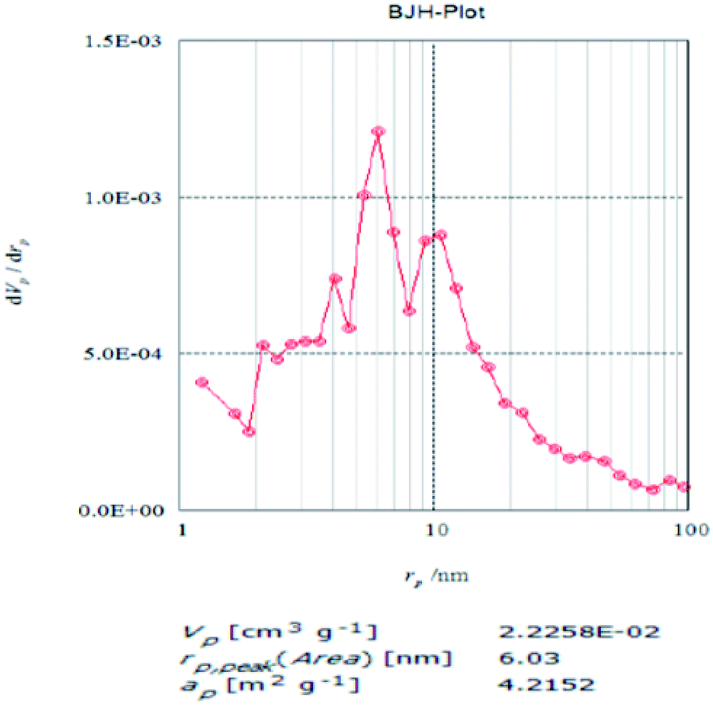
BET analysis. The BET surface area analysis (Fig. 6 and 7) generated a loop like graph upon application of the nitrogen absorption and desorption technique. The loop is evidence for the porosity of the MFA. When pressurized N2 was applied, the nitrogen was absorbed readily and as soon as the pressure was released, nitrogen was released slowly after becoming entrapped in the pores of the catalyst. The pore size distribution curve (Fig. 8) shows that the catalyst pore radii size is 6 nm, therefore belonging to the class of mesoporous compounds.
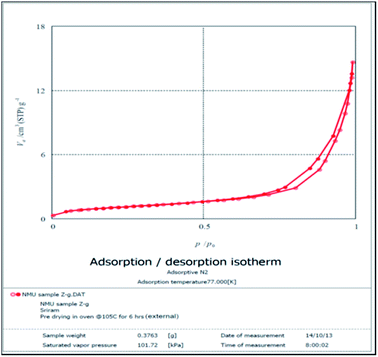 |
| | Fig. 6 BET N2 absorption/desorption isotherm. | |
 |
| | Fig. 7 BET particle size distribution graph. | |
 |
| | Fig. 8 BET pore size distribution graph. | |
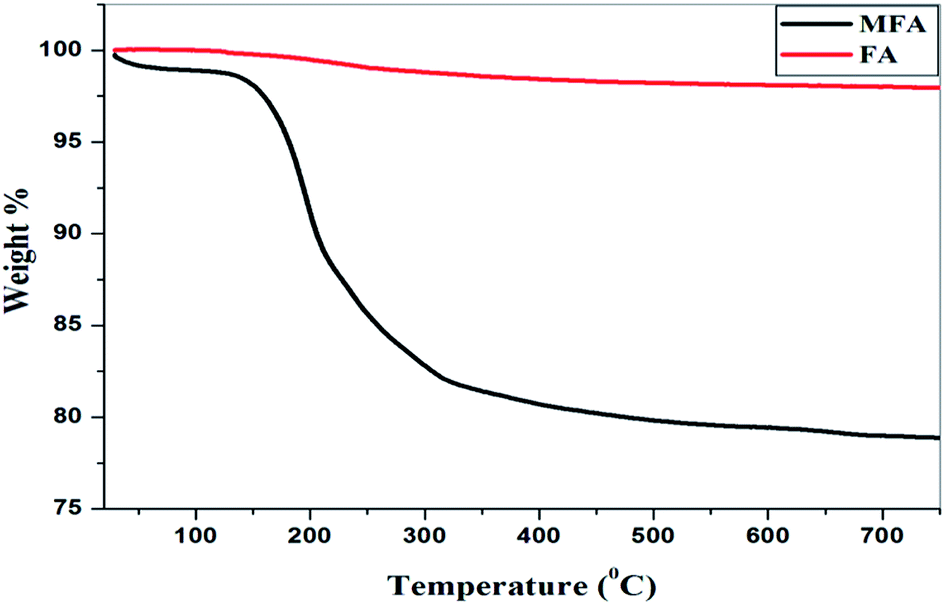
TGA analysis. The thermogravimetric analysis (Fig. 9) showed no degradation of the original fly ash sample up to 800 °C, while the MFA showed two degradation steps. The first degradation occurred between 50–100 °C due to absorbance of moisture from the surrounding humid atmosphere. The second degradation stage occurred between 180–300 °C due to breakage of the highly acidic Fe–O bond resulting in the formation of iron oxide and silica as the crystalline product decomposed into amorphous powders, which were then stable up to 800 °C. During the second degradation 20% weight loss was observed.
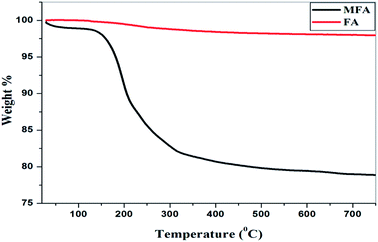 |
| | Fig. 9 Thermal gravimetric analysis. | |
XPS analysis. The X-ray photoelectronic spectroscopic analysis of MFA depicts the presence of vanadium and cerium metal impurities along with iron, aluminium, silicon, oxygen and carbon, as desired(Fig. 10). The iron that is incorporated shows a binding energy of 716 eV for its valence electron, which is far more than the binding energies in its oxide forms, like FeO at 709.8 eV and Fe2O3 at 710.8 eV, which is ultimately evidence of its tetrahedral binding with silicon through oxygen bridges. The same can be observed in case of silicon which also shows a higher binding energy than its natural oxide SiO2.
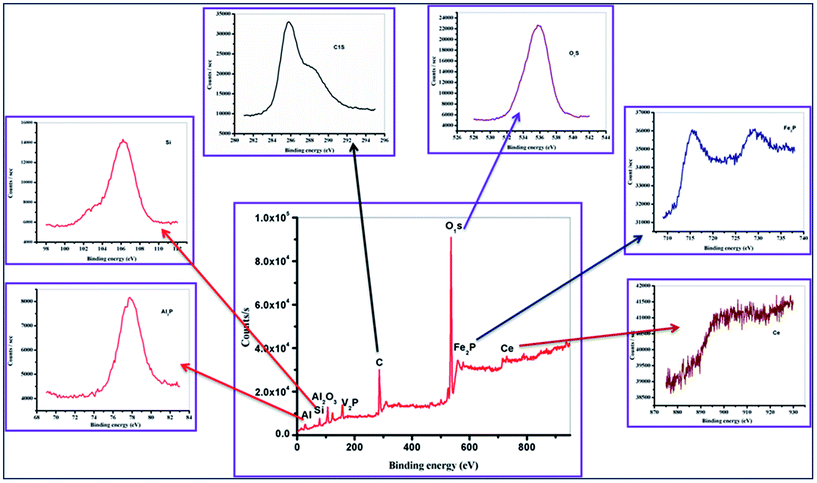 |
| | Fig. 10 X-ray photoelectronic spectroscopic analysis. | |
The time required for the reaction and the yield of the products after adding the aldehyde derivatives are summarized in Table 1. The results show that EDGs at the 2 and 4 positions enhance the rate of reaction with respect to time while in the case of bulky substituents, there is a retardation in the rate of reaction which can be attributed to the steric hindrance.
Table 1 List of synthesized azomethine derivatives of INHa
| Entry |
Code |
Ar |
H/R |
Product |
Time (min) |
Yield (%) |
Melting point (°C) |
Reported melting point (°C) |
| Note: the table contains the list of synthesized azomethine derivatives of INH with the required time for the synthesis, their yields and melting points. |
| 1 |
3a |
C6H5– |
H |
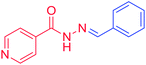 |
5 |
89 |
204–206 |
205–208 (ref. 19) |
| 2 |
3b |
p–OH–C6H5– |
H |
 |
5 |
92 |
190–192 |
189–191 (ref. 19) |
| 3 |
3c |
o–OH–C6H5– |
H |
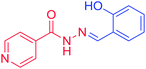 |
7 |
93 |
202–204 |
203–205 (ref. 19) |
| 4 |
3d |
m–OH–C6H5– |
H |
 |
10 |
88 |
>250 |
— |
| 5 |
3e |
o–NO2–C6H5– |
H |
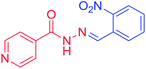 |
3 |
92 |
226–228 |
— |
| 6 |
3f |
p–NO2–C6H5– |
H |
 |
6 |
90 |
240–242 |
240–242 (ref. 20) |
| 7 |
3g |
C14H9– |
H |
 |
15 |
85 |
>250 |
— |
| 8 |
3h |
C8H6N– |
H |
 |
15 |
84 |
142–144 |
— |
The synthesized MFA has proven to be an energy efficient catalyst for the synthesis of azomethine derivatives of INH as it acts as a heterogeneous catalyst, reducing the reaction time as mentioned in earlier reports. The MFA has a porous crystalline nature and comprises of a ferrous silicate constituent in which the iron and silicon are connected by oxygen bridges just like a zeolite. The replacement of a homogeneous acid catalyst by a solid acid catalyst is the main achievement of the present study, as summarised in Table 2.
Table 2 Comparison study of catalystsa
| Entry |
Catalyst |
Time |
Temperature (°C) |
Yield (%) |
| Note: the table for comparison of the catalytic activity of MFA with parent fly ash and other reported catalysts. |
| 1 |
No catalyst20 |
2–2.5 hour |
Refluxed in EtOH |
82% |
| 2 |
Acetic acid18 |
3–4 hour |
Refluxed in EtOH |
82% |
| 3 |
Fly ash |
2–3 hour |
Refluxed in EtOH |
85% |
| 4 |
MFA |
10–30 min |
RT |
89% |
Characterization of the synthesized derivatives
3a. (E)-N′-benzylidene isonicotinohydrazide, melting point = 204–206 °C, m/z = mass analysis, base peak = M + 1, 1H NMR (DMSO, 400 MHz), δH = 1H (S 12.09), 2H (dd 8.76, 5.6 Hz, 16 Hz, 4 Hz) 1H (S 8.47), 2H (dd 7.83, 2.4 Hz, 2.8 Hz, 1.6 Hz) 2H (dd 7.75, 2.8 Hz, 5.2 Hz, 1.6 Hz) 3H (m 7.49, 0.4 Hz, 3.2 Hz, 5.2 Hz, 4.4 Hz, 2 Hz, 3.2 Hz, 1.6 Hz), 13C NMR (DMSO, 100 MHz) δC = 162.09, 150.81, 149.49, 140.93, 134.48, 130.87, 129.37, 127.74, 122.
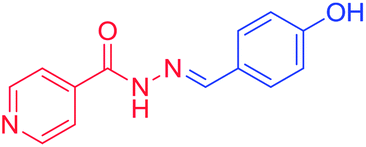
3b. (E)-N′-(4-hydroxybenzylidene) isonicotinohydrazide, melting point = 190–192 °C, m/z = mass analysis, base peak = M + 1, 1H NMR (DMSO, 400 MHz), δH = 1H (S 11.80), 1H (S 9.88), 2H (broad S 8.76), 1H (S 8.37), 2H (d 7.83, 3.68 Hz), 2H (d 8.22), 2H (d 7.85, 8.56 Hz), 2H (dd 6.80, 8.52 Hz, 8.36 Hz, 17.84 Hz), 13C NMR (DMSO, 100 MHz) δC = 161.25, 159.68, 150.04, 149.36, 140.55, 128.94, 128.44, 124.85, 121.40, 115.59.
3c. (E)-N′-(2-hydroxybenzylidene) isonicotinohydrazide, melting point = 202–204 °C, m/z = mass analysis, base peak = M + 1, 1H NMR (DMSO, 400 MHz), δH = 1H (S 12.29), 1H (S 11.17), 2H (dd 8.75, 5.64 Hz, 4.68 Hz, 18.12 Hz), 1H (S 8.67), 2H (d 7.85, 4.6 Hz, 1.32 Hz), 1H (m 7.54, 1.2 Hz, 1.2 Hz, 6.44 Hz), 1H (m 7.29, 7 Hz, 7.04 Hz, 1.48 Hz), 2H (dd 6.92, 8 Hz, 7.44 Hz, 7.12 Hz), 13C NMR (DMSO, 100 MHz) δC = 161.16, 157.62, 150.17, 149.47, 139.79, 131.46, 129.58, 121.37, 119.15, 118.32, 116.37.
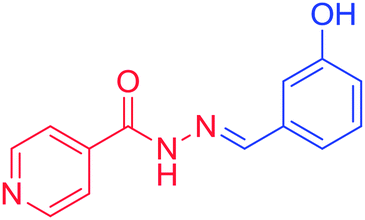
3d. (E)-N′-(3-hydroxybenzylidene) isonicotinohydrazide, melting point = more than 250 °C, m/z = mass analysis, base peak = M + 1, HRMS analysis, C13H12O2N3 = 242.0924, base peak = 242.0928, 1H NMR (DMSO, 400 MHz), δH = 1H (S 11.99), 1H (S 9.57), 2H (d 8.76, 4.2 Hz), 1H (S 8.39), 2H (d 7.23, 3.2 Hz, 8.2 Hz), 1H (d 7.12, 7.6 Hz), 1H (d 6.86, 1.64 Hz), 1H (d 6.84, 1.72 Hz), 13C NMR (DMSO, 100 MHz) δC = 162.04, 158.17, 150.8, 149.57, 140.95, 135.74, 130.43, 121.99, 119.49, 118.23, 113.18.
3e. (E)-N′-(2-nitrobenzylidene) isonicotinohydrazide, melting point = 226–228 °C, m/z = 270, mass analysis, base peak = M + 1, C13H11O3N4 = 271.0826, base peak = 271.0832, 1H NMR (DMSO, 400 MHz), δH = 1H (S 12.39), 1H (S 8.95), 2H (d 8.78, 5.76 Hz), 1H (t 8.19, 3.76 Hz, 4.12 Hz), 1H (d, 8.08, 8 Hz), 2H (d 7.86, 5.84 Hz), 2H (d 7.80, 7.56 Hz, 7.64 Hz), 1H (d 7.69, 6.24 Hz), 1H (7.65, 0.92 Hz), 13C NMR (DMSO, 100 MHz) δC = 161.76, 150.12, 148.17, 144.16, 139.9, 133.49, 130.64, 128.08, 124.48, 121.42.
3f. (E)-N′-(4-nitrobenzylidene) isonicotinohydrazide, melting point = 240–242 °C, m/z = 270, mass analysis, base peak = M + 1, 1H NMR (DMSO, 400 MHz), δH = 1H (S 12.36), 2H (broad S 8.81), 1H (S 8.59), 1H (d 8.3), 1H (d 8.22), 2H (d 8.03), 2H (d 7.86).
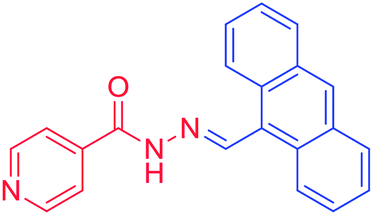
3g. (E)-N′-(anthracene-9-ylmethylene) isonicotinohydrazide, melting point = more than 250 °C, m/z = 325, mass analysis, base peak = M + 1, HRMS analysis, C21H16ON3 = 326.1288, base peak = 326.1295, 1H NMR (DMSO, 400 MHz), δH = 1H (S 12.28), 1H (S 9.70), 2H (d 8.32, 5.6 Hz), 2H (d 8.74, 8.84 Hz), 1H (S 8.65), 2H (t 8.13, 9.64 Hz, 8.28 Hz), 2H (d 7.95, 5.88 Hz), 2H (t 7.62, 6.84 Hz, 7.6 Hz), 2H (t 7.55, 7.6 Hz, 7 Hz), 13C NMR (DMSO, 100 MHz) δC = 161.47, 150.15, 148.15, 140.36, 130.84, 129.73, 128.81, 126.93, 125.27, 124.83, 124.74, 121.39.
3h. (E)-N′-((1H-indole-3-yl) methylene) isonicotinohydrazide, melting point = 142–144 °C, m/z = 264, mass analysis, base peak = M + Na, M + 1 (87%), HRMS analysis, C15H13ON4 = 265.1084, base peak = 265.1089, 1H NMR (DMSO, 400 MHz), δH = 1H (S 11.78), 1H (S 11.67), 2H (d 8.79, 4.4 Hz), 1H (S 8.65), 1H (d 8.30, 7.56 Hz, 7.12 Hz), 3H (dd 7.87, 4.76 Hz, 10.64 Hz), 1H (d 7.47, 7.76 Hz) 2H (m 7.19, 6.88 Hz, 7.76 Hz, 5.32 Hz, 7.56 Hz, 7.08 Hz, 7.68 Hz.
Conclusion
In the present work, the modification of fly ash by the incorporation of iron into its pozzolanic framework on calcination proved a key step for the development of a new heterogeneous catalyst (MFA). The synthesized MFA was confirmed as a very efficient catalyst as it has an ability to allow the reactants to react in its pore voids. The MFA, by virtue of its acid sites, polarizes the isonicotinohydrazide thereby reducing the Ea and the reaction occurs at RT in less time when compared to the conventional methods. The synthesis of azomethine derivatives of INH utilizes a green chemical pathway with a shorter reaction time, energy efficiency, ease of separation and replacement of harmful acids by a heterogeneous solid acid catalyst.
Acknowledgements
We are thankful to UGC, New Delhi for financial support through the UGC R&D Project. We thank SAIF, Chandigarh, Punjab University for spectral data. We acknowledge support of Mr Sachin Avhad, Territory Manager, Metrohelm India Limited, Navi Mumbai – 400 710 (MS) India for BET surface area and porosity analysis and characterization and CIF, UICT, NMU, Jalgaon for helping in terms of physical characterizations.
References
-
(a) V. Escande, A. Velati, C. Garel, B.-L. Renard, E. Petit and C. Grison, Green Chem., 2015, 17, 2188–2199 RSC;
(b) I. Zohar, R. Bookman, N. Levin, H. de Stigter and N. Teutsch, Environ. Sci. Technol., 2014, 48, 13592–13600 CrossRef CAS PubMed;
(c) R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
-
(a) O. Cheung and N. Hedin, RSC Adv., 2014, 4, 14480–14494 RSC;
(b) R. Chal, C. Gerardin, M. Bulut and S. V. Donk, ChemCatChem, 2013, 3, 67–81 CrossRef PubMed.
-
(a) H. L. Ngo, Lipid Technol., 2014, 26, 11–12 CrossRef PubMed;
(b) J. H. Clark, Acc. Chem. Res., 2002, 35, 791–797 CrossRef CAS PubMed.
-
(a) A. Takagaki, J. C. Jung and S. Hayashi, RSC Adv., 2014, 4, 43785–43791 CAS;
(b) L. Wang, S. Yamamoto, S. Malwadkar, S.-I. Nagamatsu, T. Sasaki, K. Hayashizaki, M. Tada and Y. Iwasawa, ChemCatChem, 2013, 5, 2203–2206 CrossRef CAS PubMed;
(c) D. Perra, N. Drenchev, K. Chakarova, M. G. Cutrufello and K. Hadjiivanov, RSC Adv., 2014, 4, 56183–56187 RSC.
- R. Y. Brogaard, C.-M. Wang and F. Studt, ACS Catal., 2014, 4, 4504–4509 CrossRef CAS.
- M. Malhotra, G. Sharma and A. Deep, Acta Pol. Pharm., 2012, 69(4), 637–644 CAS.
- M. Hermanek, R. Zboril, I. Medrik, J. Pechousek and C. Gregor, J. Am. Chem. Soc., 2007, 129, 10929–10936 CrossRef CAS PubMed.
- X. Querol, N. Moreno, J. C. Umaña, A. Alastuey, E. Hernández, A. López-Soler and F. Plana, Int. J. Coal Geol., 2002, 50, 413–423 CrossRef CAS.
- Y.-Q. Jin, X.-J. Ma, X.-G. Jiang, H.-M. Liu, X.-D. Li, J.-H. Yan and K.-F. Cen, Energy Fuels, 2013, 27, 394–400 CrossRef CAS.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., 2010, 49, 3120–3145 CrossRef CAS PubMed.
- X. Querol, N. Moreno, A. Alastuey, R. Juan, J. M. Andres, A. Lopez-soler, C. Ayora, A. Medinaceli and A. Valero, Geol. Acta, 2007, 5(1), 49–57 CAS.
- K. Li, J. Valla and J. G. Martinez, ChemCatChem, 2014, 6, 46–66 CrossRef CAS PubMed.
- M. Malhotra, V. Monga, S. Sharma, J. Jain, A. Samad, J. Stables and A. Deep, Med. Chem. Res., 2012, 21, 2145–2152 CrossRef CAS.
- R. R. Somani, A. G. Agrawal, P. P. Kalantri, P. S. Gavarkar and E. D. Clercq, Int. J. Drug Des. Discovery, 2011, 2(1), 353–360 CAS.
-
(a) A. P. G. Nikalje, M. Pathan, M. Ghodke and D. Rajani, Pharm. Sin., 2012, 3(4), 488–493 CAS;
(b) K. Tayade, S. K. Sahoo, B. Bondhopadhyay, V. K. Bhardwaj, N. Singh, A. Basu, R. Bendre and A. Kuwar, Biosens. Bioelectron., 2014, 61, 429–433 CrossRef CAS PubMed.
- N. Georgieve, Z. Yaneva, G. Nikolova and S. Simova, Adv. Biosci. Biotechnol., 2012, 3, 1068–1075 Search PubMed.
- S. Niazi, C. Javali, M. Paramesh and S. Shivaraja, Int. J. Pharm. Pharm. Sci., 2010, 2(3), 108–112 CAS.
- G. Nigade, P. Chavan and M. Deodhar, Med. Chem. Res., 2012, 21, 27–37 CrossRef CAS.
- M. Malhotra, V. Monga, S. Sharma, J. Jain, A. Samad, J. Stables and A. Deep, Med. Chem. Res., 2012, 21, 2145–2152 CAS.
- M. M. Heravi, V. Zadsirjan, K. Bakhtiari and F. F. Bamoharram, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 259–263 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03528g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.