
Sustainable synthesis of N-acetyllactosamine using an immobilized β-galactosidase on a tailor made porous polymer†
Antonio Aires-Trapoteab,
Aitana Tamayoc,
Juan Rubioc,
Angel Rumbero*b and
María J. Hernáiz*a
aComplutense University, Spain
bFaculty of Science, Autonoma University of Madrid, Spain
cInstituto de Cerámica y Vidrio (ICV), Consejo Superior de Investigaciones Científicas (CSIC), Spain
First published on 21st April 2015
Abstract
Porous polymer particles containing surface epoxy groups were synthesized for immobilizing β-galactosidase from Bacillus circulans. Enzyme immobilization was achieved by covalent attachment to a custom made porous polymer and the biocatalyst was characterized in terms of optimal pH and thermal stability, and its catalytic efficiency evaluated for synthesizing N-acetyllactosamine (Gal-β-(1 → 4)-GlcNAc) in different solvents and using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 molar ratio of donor (pNP-β-Gal):acceptor (GlcNAc). The reaction proceeded with high conversion rates and regioselectivity. Yields up to 60% of β-(1 → 4) with biosolvent 3 were found. Reusability assays were performed with the same reaction conditions finding that the immobilized enzyme retains about 85% of its activity after twenty batches with conversion yields above 60%. Furthermore, reaction scale up, biosolvent recovery and recycling are achieved retaining catalytic activity.
:5 molar ratio of donor (pNP-β-Gal):acceptor (GlcNAc). The reaction proceeded with high conversion rates and regioselectivity. Yields up to 60% of β-(1 → 4) with biosolvent 3 were found. Reusability assays were performed with the same reaction conditions finding that the immobilized enzyme retains about 85% of its activity after twenty batches with conversion yields above 60%. Furthermore, reaction scale up, biosolvent recovery and recycling are achieved retaining catalytic activity.
Introduction
Oligosaccharides are involved in a wide range of biological processes including bacterial and viral infections, cancer metastasis, the blood-clotting cascade and many other crucial intercellular recognition events.1–4 As the understanding of these biological functions increases, the need for practical synthetic procedures to obtain oligosaccharides in large quantities has become a major issue. Organic chemical methods have been developed, but they involve several elaborate protection and deprotection steps.5–7 The use of enzymes allows to overcome this burden and to this day many useful methods to obtain oligosaccharides have been reported.8–10 In fact, using enzymes as catalysts generally means a decrease in the number of reaction steps and in the waste production as well as milder reaction conditions. As a result, their production is more environmentally friendly and economically attractive and their use industry is becoming widespread.11It is well known that glycosidases are widely used enzymes that catalyse the synthesis of oligosaccharides in a kinetically controlled reaction, where a glycosyl donor is used to transfer its glycosyl residue to a sugar acceptor present in the reaction medium.12 In our research group, we have reported the regioselective synthesis of disaccharides using glycosidases in green solvents with high conversion rates and excellent regioselectivity.13–15 However, the use of glycosidases for industrial applications in organic synthesis is restricted by its relatively high cost and availability. This could be overcome by immobilization in solid supports, which allows stabilization under reaction conditions and repetitive use.16
Furthermore, immobilization of enzymes on supports, in particular on a porous support may increase the enzyme stability by preventing intermolecular processes proteolysis, aggregation, enzyme inhibition, interactions with external interfaces (stirring, pH, etc.) and allow activity determination under harsh conditions.17–22
Commercial β-galactosidase from Bacillus circulans (Biolacta N-5, Daiwa kasei) is an interesting enzyme that has already been used for the enzymatic synthesis of several β-D-(1 → 4) galactooligosaccharides bearing a GlcNAc residue at the reducing end, although some β-D-(1 → 6) linkages were also formed.24 It has been reported that this commercial β-galactosidase consists of at least a mixture of four isoforms, but the so-called β-galactosidase-I is the most abundant. This form has a monomeric structure with a molecular weight of 212 kDa and displays optimal pH in the 5.5–6.5 range.23 Despite the fact that enzymes are very efficient because of their high regio- and stereoselectivity, they have not been often considered for large-scale preparative applications. Among the known immobilization methods, covalent attachment to solid supports generally leads to very stable biocatalysts. Several supports have been tested for covalent immobilization of B. circulans β-galactosidase including silica,25–27 chitosan particles,28 polymeric membranes,29,30 methacrylate support31–33 and glyoxyl-agarose34 and many of these catalysts have been tested for the synthesis of galacto-oligosaccharides.24,25,32–34
In a previous paper, we tested the immobilization process of this β-galactosidase onto a commercial epoxy-activated support (Eupergit)31 but the stability found was not appropriate for useful applications, so further improvements were required. The epoxy groups in these supports are attached to their surface through short spacer arms, forming a very dense monolayer of reactive moieties. In this case, it is generally accepted that the immobilization of each enzyme molecule occurs through several residues, provided that the reactive groups in the support are secluded from its surface through short spacer arms.35 In this way, all the enzyme residues involved in immobilization preserve their relative position being almost completely unaffected during any conformational change that may occur promoted by heat, organic solvents or any other distorting agent. For this reason, highly activated epoxy polymers are good supports to have stabilizing multi-point-immobilization sites.35–39
Covalent immobilization onto Eupergit or any activated epoxy polymer follows a two-step mechanism. The primary event is a rapid physical adsorption of the protein onto the resin; in a second step, a covalent reaction takes place between the adsorbed protein and the support. Epoxy groups react with different groups (amino or thiol groups) in proteins under very mild experimental conditions (e.g. pH 7.5), with minimal chemical modification of the protein and formation of very stable secondary amine bonds.40,41
As part of our ongoing research focused on biocatalytic synthesis of disaccharides with biological interest, we report here a new immobilized β-galactosidase on a tailor made porous polymer to produce N-acetyllactosamine using biosolvents (solvents derived from biomass). To the best of our knowledge, this is the first study of covalent immobilization of β-galactosidase from B. circulans in this kind of support. Once the transglycosidation reaction was completed, biosolvents were separated and the system could be reused in several cycles.
Results and discussion
Synthesis of porous epoxy-activated polymers
Porous epoxy-activated polymers were synthesized by a standard solution polymerization technique.42,43 A reaction mixture composed of allyl glycidyl ether (AGE) as monomer and divinyl benzene (DVB) as crosslinker was used. The porosity of these polymers can be controlled varying several parameters such as: porogenic solvent,44 percentage of monomer and crosslinker,45 percentage of initiator and polymerization temperature.46 Cyclohexanol, dodecanol and toluene were tested as porogenic solvents. However, only cyclohexanol gave polymers with large size pores. The factors that most influence the immobilization mechanism of enzymes are pore size and surface area.18–22,47 Therefore it is important to have a large pore size to facilitate the accessibility of the enzyme inside the pores and also to avoid substrate diffusion problems into the enzyme. At the same time, it is desirable to have supports with the maximum surface-area ratio to facilitate the immobilization of large amounts of enzyme and minimize the catalysts volume, thus obtaining an immobilized enzyme with high activity per gram of support. Poly(AGE-co-DVB)-27 (Table S1 in ESI† summarizes the results obtained varying polymerization parameters) was selected as the copolymer with best pore size/surface area ratio, to carry out the immobilization-stabilization procedure of Bacillus circulans β-galactosidase. The chemical, morphological and structural characterization of the poly(AGE-co-DVB)-27 was performed using standard techniques.Characterization of poly(AGE-co-DVB)-27
 | ||
| Fig. 1 FT-IR spectrum of AGE and poly(AGE-co-DVB)-27. | ||
The FT-IR spectra show the typical stretching vibrations of epoxy groups at 1256 and 903 cm−1 clearly indicating their presence in the copolymers. On the other hand, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching vibrations respectively at 1650 and 1635 cm−1, confirm the presence of ester groups in the copolymer structure coming from the AGE monomer.
O and C–O stretching vibrations respectively at 1650 and 1635 cm−1, confirm the presence of ester groups in the copolymer structure coming from the AGE monomer.
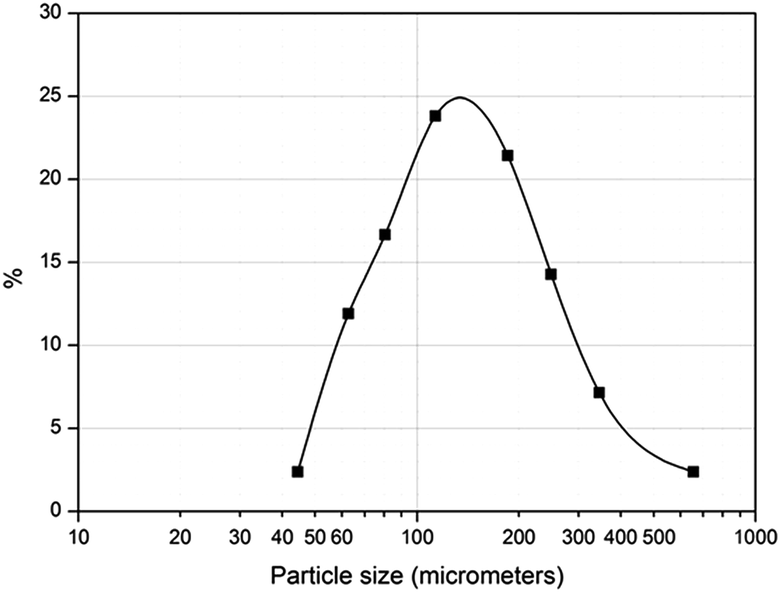 | ||
| Fig. 2 Particle size distribution (PSD) of poly(AGE-co-DVB)-27. | ||
 | ||
| Fig. 3 FE-SEM photographs showing the microstructure of porous of poly(AGE-co-DVB)-27. | ||
According to these images it is clear that the polymer particles consist of an aggregate of nanoparticles with an interconnected porous network. Most of the pores are in the range of macroporosity, i.e., higher than 50–100 nm and therefore they are suitable for solution diffusion without any pressure drop. Thus, enzyme immobilization as well as washing and solution removing experiments can be carried out in normal conditions and therefore polymer reusability and recycling are possible.
Enzyme immobilization
We have previously used β-D-galactosidase from Bacillus circulans to synthesize disaccharides in biosolvents,49,50 with excellent yields and high regioselectivity. We have also reported the immobilization of β-D-galactosidase by covalent binding to Eupergit C,31 although the low reusability (best derivatives were only recycled four times) of these immobilized biocatalysts would hamper any kind of scaling up protocol.Thus, we have carried out the first covalent immobilization of commercial β-D-galactosidase from Bacillus circulans. In these study, the poly(AGE-co-DVB)-27 support was tested and the results previously obtained with commercial Eupergit C and Eupergit C 250 were used for comparative purposes.31 Eupergit C is a spherical acrylic polymer (surface area: 45 m2 g−1; pore size: 10 nm; pore volume: 0.06 mL g−1), made by copolymerization of N,N-methylene-bis(methacrylamide), glycidyl methacrylate, allyl glycidyl ether, and methacrylamide. This epoxide-containing copolymer (0.6 mmol epoxy groups per g dry weight,51 is particularly suitable for covalent immobilization of enzymes for industrial applications because of its reactor-compatibility.52 The copolymer described in this paper, poly(AGE-co-DVB)-27, surface area: 95 m2 g−1; pore size: 25 nm; pore volume: 0.6 mL g−1), also contains epoxide groups as the reactive components, but displays a higher density of reactive epoxy groups (2 mmol epoxy groups per g dry weight) compared to Eupergit C.
The enzyme load during the immobilization step was set to 200 mg of enzyme preparation (10% of protein, 20 mg of protein) for 400 mg of polymer in 40 mL of buffer (0.05 M Tris/HCl pH 7.3 buffer, 0.5 M NaCl). These conditions were selected according to previously described studies, where the same enzyme was immobilized onto a commercial support functionalized with Eupergit C.31 In these experiments, the commercial preparation of the enzyme was incubated in the presence of Eupergit C at concentrations of various ionic strengths and it was found that 0.5 M of NaCl was the best salt concentration for the adsorption of protein on the support. This result corroborates previous studies on protein adsorption on supports where high ionic strengths are needed.31,37,38
The enzyme load on poly(AGE-co-DVB)-27 was 4.8% (w/w) similar to previous protein binding loads with Eupergit C and Eupergit 250 L (commercially available epoxy polymers).31 On the other hand, increased activity was found when poly(AGE-co-DVB)-27 was used (85%), compared to Eupergit C and Eupergit 250 (67% and 53%). Based on these results, all the following experiments were conducted with the immobilized derivative, Biolacta-poly(AGE-co-DVB)-27.
Effect of pH and thermal stability on enzyme activity
The effect of pH on the activity of free and Biolacta-poly(AGE-co-DVB)-27 was performed in a 4 to 7 pH range from. The enzymatic activity of free and immobilized enzyme was measured monitoring p-nitrophenol from p-nitrophenyl β-D-galactopyranoside hydrolysis, after keeping the reaction mixture in a shaking water bath at 37 °C for 45 min in order to equilibrate the internal pH. The results are presented in Fig. 4A. As it can be seen, the Biolacta-poly(AGE-co-DVB)-27 exhibited a shift in the optimal pH of about 0.5 units towards acidic pH values, indicative of the activated matrix behaving as a polycation. Immobilization process affected the pH profile of enzyme activity (maximum at pH 5.5 for immobilized enzyme and pH 6 for free enzyme), presumably due to pKa changes of functionally essential residues that interact with the polymer environment.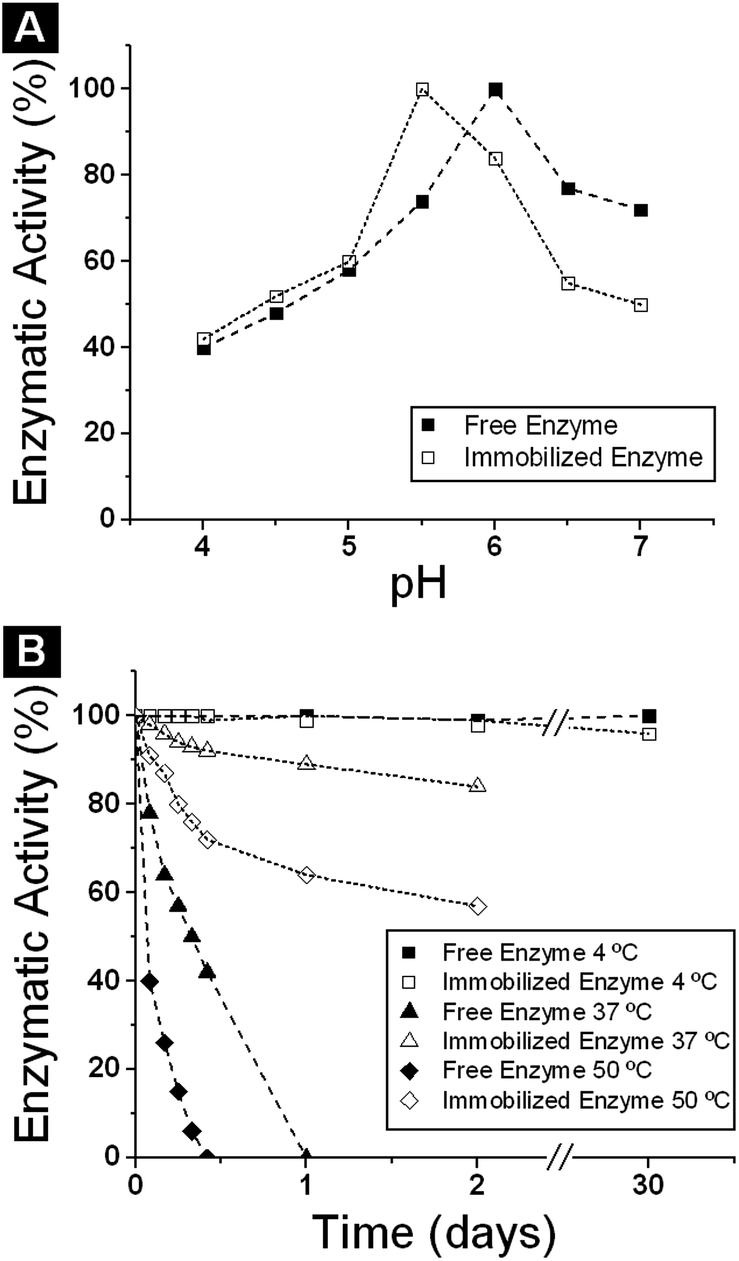 | ||
| Fig. 4 (A) Effect of pH on the activity of free and immobilized β-galactosidase from Bacillus circulans. (B) Thermal stability of free and immobilized Biolacta β-galactosidase from Bacillus circulans at 4 °C, 37 °C and 50 °C. | ||
Similar results have been reported for this enzyme immobilized on Eupergit C31,32 and different enzymes immobilized on the same support, such as pectinlyase,53 or lipases.54 Eupergit C and Eupergit C 250 L derivatives displayed better stabilities in the 5.0–6.5 range, with maximum stability at pH 5.5.31,32
The immobilized enzyme preparations can be stored at 4 °C for at least one month without appreciable deactivation (less than 5%). Thermal stability experiments were performed as described in the Experimental section, and Fig. 4B shows the thermal stability behaviour of the commercial and immobilized enzymes.
Comparison of the stability of the soluble enzyme and the enzyme derivatives revealed that covalent immobilization, followed by blocking, provides benefits in terms of thermal stability of the insoluble biocatalysts (Fig. 4). In fact, after 24 h at 37 °C the residual activity of poly(AGE-co-DVB)-27 derivative blocked with glycine was about 95% compared with 0% for the corresponding free enzyme. At 50 °C the behaviour of the free and immobilized enzyme was similar but in this case the poly(AGE-co-DVB)-27 derivative showed 60% of the enzymatic activity and the free enzyme 0%. In all cases, the poly(AGE-co-DVB)-27 derivatives exhibited greater thermostability than the free enzyme. The mild hydrophobic character of the acrylic supports favours hydrophobic interactions between support and protein surface, and this may affect the enzyme properties. It has been suggested that glycine blocking generates a hydrophilic microenvironment that favours enzyme stability.39 This enzyme stabilization could be explained by the multipoint covalent attachment in these biocatalysts.32,55
Transglycosylation reactions in buffer and biosolvents
The β-galactosidase from Bacillus circulans (Biolacta) is a valuable biocatalyst for galactosyl transfer from suitable donors onto a variety of substrates.56 With N-acetyl glucosamine or N-acetyl-glucosamine glycosides as acceptors, β-(1 → 4) transfer product predominates giving N-acetyl lactosamine, but some β-(1 → 6) galactosyl transfer is also observed (Scheme 1).55 | ||
| Scheme 1 General scheme of transglycosylation reaction catalyzed by β-galactosidase from Bacillus circulans (Biolacta). | ||
The influence of the immobilization of the β-galactosidase from B. circulans (Biolacta) on poly(AGE-co-DVB)-27 copolymer on transglycosylation reactions was studied. Fig. 5 shows the transglycosylation profile of B. circulans β-D-galactosidase and Biolacta-poly(AGE-co-DVB)-27 derivative with p-nitrophenyl-β-D-galactopyranoside (pNP-Gal) as donor and GlcNAc as acceptor.
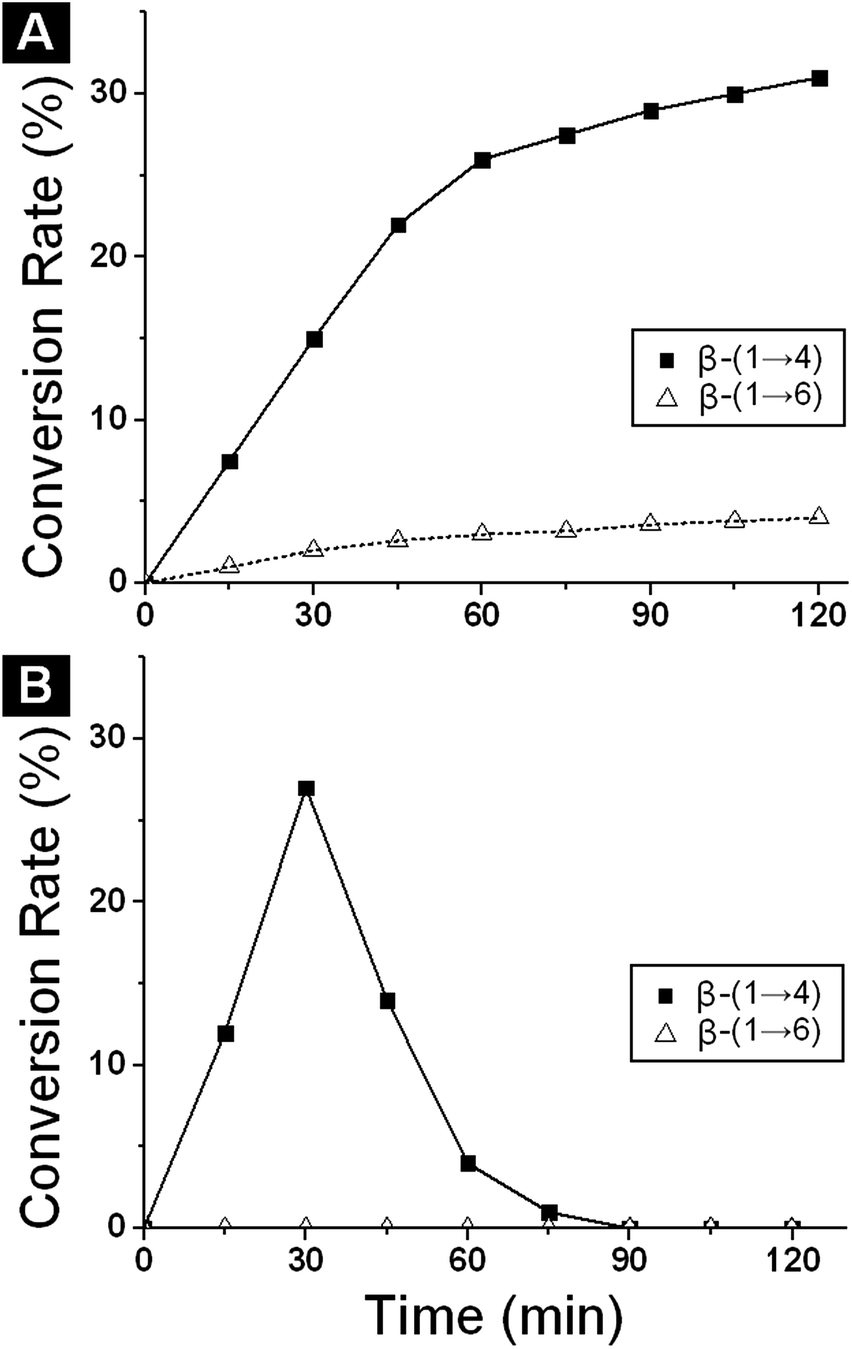 | ||
| Fig. 5 Time course of the formation of N-acetyl lactosamine and of its β(1 → 6) isomer by the action of the β-galactosidase from Bacillus circulans: (A) free enzyme; (B) immobilized enzyme. | ||
The amount of N-acetyllactosamine (β-(1 → 4) isomer) and its β-(1 → 6) isomer produced as a function of time was examined. A key objective was to compare the regioselectivity of the free and immobilized enzyme, which could vary upon immobilization. The amounts of N-acetyllactosamine and its β-(1 → 6) isomer were examined as a function of time, and samples were analysed by HPLC. With the free enzyme, production of N-acetyl lactosamine reached a maximum of 30% at 120 min at which point 5% of the β-(1 → 6) isomer was present. Formation of the 1 → 6 isomer was much slower than formation of the β(1 → 4) isomer (Fig. 5A). In the case of the immobilized enzyme in buffer, similar conversion was achieved (27% N-acetyl lactosamine) but at shorter reaction time (30 min) and more regioselectivity (none of the β(1 → 6) isomer was detected) than with free enzyme. This suggests that the immobilization process produced a change in the behaviour of the enzyme. As we can see in Fig. 5B, after 30 min, a significant decrease of the product concentration due to enzymatic hydrolysis was observed. This is a result of the reaction being under kinetic control and yields depend on the enzyme. They are reach a maximum after 30 minutes and at this point the product becomes a substrate and it is over equilibrium concentration.
We have already shown, that the use of certain biosolvents (biomass derived solvents) in the reaction medium increases the activity and direct the regioselectivity of the free β-galactosidase from B. circulans in transglycosylation reactions obtaining only the β(1 → 6) regioisomer in excellent yields.49,50 Based on these results, solvents 1, 2 and 3 (Scheme 2) were selected for further studies with the Biolacta-poly(AGE-co-DVB)-27 derivative.
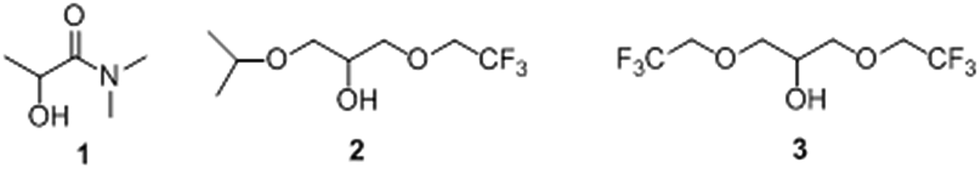 | ||
| Scheme 2 Structure of biosolvents derived from biomass. | ||
Transglycosylation reactions catalysed by immobilized β-galactosidase from B. circulans (Biolacta-poly(AGE-co-DVB)-27) were carried out following the general procedure described in the Experimental section and monitored by HPLC. The concentration of biosolvent was set to 2 M in the mixture and buffered with a 100 mM sodium phosphate at pH 6.0.49,50 Formation of N-acetyllactosamine and β-(1 → 6) isomer were examined as a function of time. The results obtained for the transglycosylation reaction with the immobilized β-galactosidase in the presence of different biosolvents are summarized in Fig. 6 and Table 1.
 | ||
| Fig. 6 Time course of the formation of N-acetyllactosamine catalyzed by β-galactosidase from Bacillus circulans immobilized on poly-(AGE-co-DVB)-27 and using sodium phosphate buffer pH 6.0 and 2 M of biosolvent. | ||
| Solvent | β-(1 → 4) (%) | Time (min) | Productivity (mmol h−1 mg−1) | |
|---|---|---|---|---|
| Free enzyme | Buffer | 31 | 120 | 1.1 × 10−3 |
| Immobilized enzyme | Buffer | 30 | 30 | 4.1 × 10−3 |
| 1 | 15 | 15 | 4.1 × 10−3 | |
| 2 | 60 | 90 | 2.8 × 10−3 | |
| 3 | 60 | 15 | 1.7 × 10−2 |
In the presence of solvents 1, 2 and 3 the regioisomer β-(1 → 4) was formed as the sole product and maximum conversions (60%) were reached with solvent 2 after 15 min and solvent 3 after 90 min. In all cases, none of the β-(1 → 6) isomer was detected, clearly indicating that the immobilization procedure did not alter the regioselectivity of the β-galactosidase in the presence of biosolvents presumably due to enzyme structural rigidification after enzyme immobilization. Most likely, blocking conformational changes of the enzyme that favour the disposition of the substrates in the active center for the formation of β-(1 → 4) linkage. In addition a significant increase of conversion compared to free enzyme in buffer was observed. In the case of solvent 2, the reaction was slower, and after 90 min a 60% of N-acetyllactosamine was obtained. For solvent 3 the time of reaction was reduced (60% yield at 15 minutes). For this reason, this immobilized enzyme in buffer shows higher catalytic activity than free enzyme (1.0 × 10−3 mmol h−1 mg−1 for free enzyme and 4.1 × 10−3 mmol h−1 mg−1 for immobilized enzyme) and the presence of biosolvent 3 also increases this productivity (4.1 × 10−3 mmol h−1 mg−1 in buffer and 1.7 × 10−2 mmol h−1 mg−1 in 2 M of solvent 3).
Thus, pure N-acetyllactosamine was easily isolated by chromatography on a column of charcoal–celite and structural determination was done by 1H-NMR and 13C-NMR. Spectra were identical to the ones previously reported.57
Re-use of the immobilized Biolacta-poly(AGE-co-DVB)-27 in the enzymatic synthesis of N-acetyl lactosamine with biosolvents
The potential for the re-use of the supported enzyme in the synthesis of N-acetyllactosamine was investigated. After the first assay, Biolacta-poly(AGE-co-DVB)-27 was recovered, washed and re-assayed with fresh substrate mixture under the same experimental conditions, and this procedure was repeated 20 times, using the biosolvents 1, 2, 3 and buffer. The experimental results shown in Fig. 7 illustrate the excellent reusability and stability these buffered solvents for the immobilized enzyme, which retained 90–95% of its initial activity after 10th catalytic cycles and 80–85% after 20th catalytic cycles (Fig. 7). These results suggest that, over 20 reaction cycles under the above reaction conditions, no significant leaching of β-galactosidase from poly(AGE-co-DVB)-27 or denaturation of immobilized β-galactosidase occurred. Nevertheless, the loss of activity may be ascribed to the blocking of some β-galactosidase active sites or to the gradual lost of bound β-galactosidase during catalysis. | ||
| Fig. 7 Reusability of Bacillus circulans β-galactosidase (Biolacta) immobilized on poly(AGE-co-DVB) in presence of different biosolvents. | ||
It was also observed that the selectivity for N-acetyllactosamine formation remains almost unchanged after re-use. These results showed that this immobilized enzyme retained 80% of its initial activity after 20 uses and the same enzyme immobilized on Eupergit retained 80% of its initial activity only after 3 uses.56 The main advantage in the enzyme reuse in biosolvents compared to buffers, is the higher yields of N-acetyl lactosamine (60% vs. 27%) in biosolvents.
On the other hand, an advantage of solvents 2 and 3, compared to solvent 1 used in this study, is that under these experimental conditions there is a biphasic mixture between these solvents and aqueous buffer,58 and then after reaction these solvents can be separated from the reaction media by centrifugation. Moreover, carbohydrate compounds in the reaction media are not soluble in this solvent phase and remain in the aqueous phase. Centrifugation becomes a very useful tool for the isolation of these solvents from the reaction media, allowing its reuse in further reactions. This clearly shows the advantage in the use of biosolvents.
Scale up of the transglycosylation reactions
Based on the excellent results obtained with the immobilized β-galactosidase, enzymatic synthesis of N-acetyllactosamine was scaled-up 10 times, up to 20 mL volume. The reaction was scaled up with solvents 2 and 3. In the case of solvent 2, a final conversion of 55% after 100 minutes was observed. Similar results were obtained with solvent 3 but in 20 minutes confirming the viability of the process scale up.Conclusions
A new porous material (poly(AGE-co-DVB)-27) with large surface areas and porous sizes has been obtained and characterized. This support was used for the immobilization of β-galactosidase from Bacillus circulans. Results show that this material allowed high enzyme loading and excellent catalytic activity of β-galactosidase, favouring its thermal stabilization at 37 and 50 °C. At the same time, the immobilized enzyme was found to be highly efficient and regioselective in the enzymatic synthesis of N-acetyllactosamine in the presence of biosolvents. The immobilized enzyme and biosolvent could be reused up to 20 cycles with excellent activity retention and regioselectivity. Optimization of the reaction conditions allows the reaction scale up to 20 mL with further solvent recycling and reuse. This strategy constitutes an excellent system for developing a successful and efficient bioprocess.Experimental section
Materials
Allylglycidyl ether (AGE), divinylbenzene (DVB), 2,2′-azobis (isobutyronitrile) (AIBN), cyclohexanol, p-nitrophenol (pNP), p-nitrophenyl-β-D-galactopyranoside (pNP-β-Gal), N-acetyl-D-glucosamine (GlcNAc), Gal-β(1 → 4)GlcNAc, Gal-β-(1 → 6)GlcNAc, D-(+)-galactose, ammonium sulphate and bovine serum albumin (BSA) were obtained from Sigma-Aldrich. Tween 80 was obtained from Acros Organics. Dye reagent for protein determination was purchased from BioRad. All other chemicals were from analytical grade. All other chemicals were from analytical grade.Commercially available β-galactosidase from Bacillus circulans (Biolacta N5) was a gift from Daiwa Kasei.
Solvents derived from glycerol were kindly donated by Prof. José I. García, solvents derived from biomass were a gift from Cognis IP Management GmbH, now part of BASF.
UV-visible spectra were recorded on a UV-2401 PC Shimadzu. HPLC Jasco with an evaporative light scattering detector (ELSD) using NH2P50-4E amino column (Asahipak, Japan) with acetonitrile:water (80:20) as a mobile phase at a flow of 1.0 mL min−1. NMR spectra were recorded on Bruker AV 500 MHz spectrometers. The structures of the enzymatically synthesized disaccharides were assigned by proton–proton shift correlation, carbon–proton shift correlation and DEPT-experiments; β(1 → 4) and β(1 → 6) linkages were identified by the marked downfield shift of the C-4 and C-6 resonances.
Methods
(A) Surface area and pore size analysis. Specific Surface Area (SSA), Pore Volume (PV) and Mean Pore Size (MPS) were determined by nitrogen gas adsorption–desorption a 77 K by using a Tri-Star 3000 instrument (Micrometrics, USA). Samples were degassed at 120 °C for 18 h. SSA values were calculated using the BET equation59 in the nitrogen partial pressure range of 0.05–0.35. PV and MPS were obtained by using the adsorption branch of the nitrogen isotherms according to the BJH method in the nitrogen partial pressure range of 0.35–0.99.60
(B) FT-IR spectroscopy. A Perkin-Elmer 681-Fourier transform infrared spectrophotometer with a resolution of 1 cm−1 in the transmission mode was used to study the infrared absorption. The synthesized polymers (2.0 mg) were milled with potassium bromide (100 mg) and pressed into a solid disk of 1.2 cm diameter prior to the infra-red measurement.
(C) Scanning electron microscopy (SEM). Surface morphology of poly(AGE-co-DVB)-27 was observed by Field-Emission Scanning Electron Microscopy (FE-SEM). Specimen preparation was done as follows: dried poly(AGE-co-DVB)-27 particles were mounted on stubs and sputter-coated with gold. Micrographs were taken on a Hitachi-S4700 FE-SEM instrument.
(D) Thermal characterization. Thermo Gravimetric Analysis (TGA) was performed using an AQ-500 TA Instruments equipment. For each essay 4–5 mg of polymer were used. The heating rate was set at 5 °C min−1 and all the experiments were carried out under a constant nitrogen flow of 20 mL min−1.
(E) Polymer epoxidation degree. Quantitative determination of epoxide groups in the polymers was carried out by chemical titration.61 The polymer (2.0 g) was re-suspended in CH2Cl2 (30 mL) and treated with a 20 wt% solution of tetraethyl ammonium bromide in glacial acetic acid, prepared previously. After addition of 6–8 drops of crystal violet indicator solution in acetic acid, the mixture was titrated with 0.1 N perchloric acid solution in acetic acid. Hydrobromic acid formed in situ by the exchange reaction between perchloric acid and tetraethylammonium bromide reacted instantaneously with the epoxide group, leading to bromohydrin formation. The end point of the titration was determined by the change of the colour of the solution from a sharp blue to green.
Thermal stability experiments were performed with free and immobilized enzymes which were incubated in the absence of substrate at 37 °C and 50 °C. The immobilized enzymes were placed in 100 mM sodium phosphate buffer (pH 6.0) and the specific enzymatic activities were measured at different storage times.
000 rpm with the aim of separating the aqueous phase (containing carbohydrate compounds) from the biosolvent. After that, aqueous phase was lyophilized to eliminate the water. Powder was loaded on activated carbon and Celite® column (50% m/m), products were eluted with milliQ water in linear gradient of ethanol (from 0% v/v to 15% v/v). Disaccharide enriched fractions were collected in 10% or 15% ethanol; samples were pooled, solvent was removed and then lyophilized, purity of the solid powder was analysed by HPLC.Notes and references
- M. E. C. Caines, H. Zhu, M. Vuckovic, L. M. Willis, S. G. Withers, W. W. Wakarchuk and N. C. J. Strynadka, J. Biol. Chem., 2008, 283, 31279–31283 CrossRef CAS PubMed.
- H. Shirato, S. Ogawa, H. Ito, T. Sato, A. Kameyama, H. Narimatsu, Z. Xiaofan, T. Miyamura, T. Wakita, K. Ishii and N. Takeda, J. Virol., 2008, 82, 10756–10767 CrossRef CAS PubMed.
- G. F. Springer, Science, 1984, 224, 1198–1206 CAS.
- G. F. Springer, P. R. Desai, W. Wise, S. C. Carlstedt, H. Tegtmeyer, R. Stein and E. F. Scanlon, Immunol. Ser., 1990, 53, 587–612 CAS.
- R. R. Schmidt and E. Rucker, Tetrahedron Lett., 1980, 21, 1421–1424 CrossRef CAS.
- R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef PubMed.
- H. Paulsen, Angew. Chem., Int. Ed. Engl., 1982, 21, 155–173 CrossRef PubMed.
- E. García-Junceda, J. F. García-Garcıía, A. Bastida and A. Fernández-Mayoralas, Bioorg. Med. Chem., 2004, 12, 1817–1834 CrossRef PubMed.
- M. Takeomi and T. Usui, Biosci., Biotechnol., Biochem., 2006, 70, 1049–1059 CrossRef PubMed.
- M. D. Alison, A. M. Beatrice and S. L. Flitsch, Curr. Opin. Chem. Biol., 2004, 8, 106–113 CrossRef PubMed.
- M. J. Hernaiz, A. R. Alcantara, J. I. Garcia and J. V. Sinisterra, Chem.–Eur. J., 2010, 16(31), 9422–9437 CrossRef CAS PubMed.
- A. Trincone and A. Giordano, Curr. Org. Chem., 2006, 10, 1163–1193 CrossRef CAS.
- M. Perez-Sanchez, A. Cortes-Cabrera, H. Garcia-Martin, J. V. Sinisterra, J. I. Garcia and M. J. Hernaiz, Tetrahedron, 2011, 67, 7708–7712 CrossRef CAS PubMed.
- M. Sandoval, A. Cortes, C. Civera, J. Trevino, E. Ferreras, M. Vaultier, J. Berenguer, P. Lozano and M. J. Hernaiz, RSC Adv., 2012, 2, 6306–6314 RSC.
- C. Bayon, A. Cortes, J. Berenguer and M. J. Hernaiz, Tetrahedron, 2013, 69, 4973 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- E. Magner, Chem. Soc. Rev., 2013, 42, 6213–6222 RSC.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262–6276 RSC.
- A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS PubMed.
- A. Vetere and S. Paoletti, Biochim. Biophys. Acta, 1998, 1380, 223–231 CrossRef CAS.
- T. Usui, S. Kubota and H. Ohi, Carbohydr. Res., 1983, 244, 315–323 CrossRef.
- Z. Mozaffar, K. Nakanishi and R. Matsuno, Appl. Microbiol. Biotechnol., 1986, 25, 224–228 CAS.
- C. Bernal, L. Sierra and M. Mesa, J. Mol. Catal. B: Enzym., 2012, 84, 166–172 CrossRef CAS PubMed.
- Z. Mozaffar, Z. Nakanishi and R. Matsuno, Biotechnol. Lett., 1988, 10, 805–808 CrossRef CAS.
- T.-C. Cheng, K.-J. Duan and D.-C. Sheu, J. Chem. Technol. Biotechnol., 2006, 81, 233–236 CrossRef CAS PubMed.
- D. Sen, A. Sarkar, A. Gosling, S. L. Gras, G. W. Stevens, S. E. Kentish, P. K. Bhattacharya, A. R. Barber and C. Bhattacharjee, J. Membr. Sci., 2011, 378, 471–478 CrossRef CAS PubMed.
- T. Palai and P. K. Bhattacharya, J. Biosci. Bioeng., 2013, 115, 668–673 CrossRef CAS PubMed.
- M. J. Hernaiz and D. H. G. Crout, Enzyme Microb. Technol., 2000, 27, 26–32 CrossRef CAS.
- P. Torres and F. Batista-Viera, J. Mol. Catal. B: Enzym., 2012, 83, 57–64 CrossRef CAS PubMed.
- P. Torres and F. Batista-Viera, J. Mol. Catal. B: Enzym., 2012, 74, 230–235 CrossRef CAS PubMed.
- P. Urrutia, C. Mateo, J. M. Guisan, L. Wilson and A. Illanes, Biochem. Eng. J., 2013, 77, 41–48 CrossRef CAS PubMed.
- C. Mateo, O. Abian, R. Fernandez-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2000, 26, 509–515 CrossRef CAS.
- E. Katchalski-Katzir and D. M. Kraemer, J. Mol. Catal. B: Enzym., 2000, 10, 157–176 CrossRef CAS.
- O. Barbosa, R. Torres, C. Ortiz, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Biomacromolecules, 2013, 14, 2433–2462 CrossRef CAS PubMed.
- C. Mateo, G. Fernández-Lorente, O. Abian, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2000, 1, 739–745 CrossRef CAS.
- C. Mateo, V. Grazú, B. C. C. Pessela, T. Montes, J. M. Palomo, R. Torres, F. López-Gallego, R. Fernández-Lafuente and J. M. Guisán, Biochem. Soc. Trans., 2007, 35, 1593–1601 CrossRef CAS PubMed.
- C. Mateo, O. Abian, G. Fernandez-Lorente, J. Pedroche, R. Fernandez-Lafuente, J. M. Guisan, A. Tam and M. Daminati, Biotechnol. Prog., 2002, 18, 629–634 CrossRef CAS PubMed.
- J. Turkova, K. Bláha, M. Malaníková, D. Vancurova, F. Svec and J. Kalal, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1978, 524, 162–169 CrossRef CAS.
- F. Svec and J. M. J. Frechet, J. Chromatogr., A, 1995, 702, 89–95 CrossRef CAS.
- P. A. Levkin, F. Svec and J. M. J. Frechet, Adv. Funct. Mater., 2009, 19, 1993–1998 CrossRef CAS PubMed.
- S. Scheler, J. Appl. Polym. Sci., 2007, 105, 3121–3131 CrossRef CAS PubMed.
- V. Sciannamea, J. M. Jerome and C. Detzembleur, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1219–1235 CrossRef CAS PubMed.
- F. Svec and J. M. J. Frechet, Macromolecules, 1995, 28, 7580–7582 CrossRef CAS.
- H. Takahashi, B. Li, T. Sasaki, C. Miyazaki, T. Kajino and S. Inagaki, Chem. Mater., 2000, 12, 3301–3305 CrossRef CAS.
- R. P. Quirk and Q. Zhuo, Macromolecules, 1997, 30, 1531–1539 CrossRef CAS.
- M. Perez-Sanchez, M. Sandoval, A. Cortes-Cabrera, H. Garcia-Marin, J. V. Sinisterra, J. I. Garcia and M. J. Hernaiz, Green Chem., 2011, 13, 2810–2817 RSC.
- M. Perez-Sanchez, M. Sandoval and M. J. Hernaiz, Tetrahedron, 2012, 68, 2141–2145 CrossRef CAS PubMed.
- A. G. de Segura, M. Alcalde, M. Yates, M. L. Rojas-Cervantes, N. Lopez-Cortes, A. Ballesteros and F. J. Plou, Biotechnol. Prog., 2004, 20(5), 1414–1420 CrossRef CAS PubMed.
- T. Boller, C. Meier and S. Menzler, Org. Process Res. Dev., 2002, 6(4), 509–519 CrossRef CAS.
- G. Spagna, P. G. Pifferi and A. Martino, J. Chem. Technol. Biotechnol., 1993, 57(4), 379–385 CrossRef CAS PubMed.
- (a) Z. Knezevic, N. Milosavic, D. Bezbradica, Z. Jakovljevic and R. Prodanovic, Biochem. Eng. J., 2006, 30(3), 269–278 CrossRef CAS PubMed; (b) C. Tecelao, M. Guillen, F. Valero and S. Ferreira-Dias, Biochem. Eng. J., 2012, 67, 104–110 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernández-Lorente, J. M. Guisán and R. Fernández-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS PubMed.
- M. J. Hernaiz and D. H. G. Crout, J. Mol. Catal. B: Enzym., 2000, 10, 403–408 CrossRef CAS.
- M. Sandoval, E. Ferreras, M. Pérez-Sánchez, J. Berenguer, J. V. Sinisterra and M. J. Hernáiz, J. Mol. Catal. B: Enzym., 2012, 74, 162–169 CrossRef CAS PubMed.
- C. Bayón, A. Cortés, A. Aires-Trapote, C. Civera and M. J. Hernáiz, RSC Adv., 2013, 3, 12155–12163 RSC.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73(1), 373–380 CrossRef CAS.
- R. P. Quirk and Q. Z. Zhuo, Macromolecules, 1997, 30(6), 1531–1539 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72(1–2), 248–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03527a |
| This journal is © The Royal Society of Chemistry 2015 |