
Recent advances in electrochemical detection of toxic Cr(VI)
Wei Jin*a and
Kai Yan*b
aMaterials Research Center, Missouri University of Science and Technology, Rolla, Missouri 65409, USA. E-mail: jinw@mst.edu; Fax: +1 573 341 6934; Tel: +1 573 341 4430
bSchool of Engineering, Brown University, Providence, RI 02912, USA. E-mail: Kai_Yan@brown.edu
First published on 7th April 2015
Abstract
Large quantities of highly toxic hexavalent chromium (Cr(VI)) compounds are often discharged from waste streams into the environment from various industrial processes. As stated by the World Health Organization (WHO), a maximum limit of 50 μg L−1 hexavalent chromium in groundwater system has been set for human and environmental concerns. Therefore, reliable and convenient monitoring of Cr(VI) is significantly important and emergent. Recently, electrochemical detection of hexavalent chromium has been proven as one of the most efficient methodologies and attracted increasing interest since it offers high sensitivity and powerful information, inherent miniaturization, cost-effectiveness and compatibility with advanced microfabrication technologies. This review comments on recent achievements in the electrode materials and detection techniques for electrochemical quantification of hexavalent chromium, and outlines key challenges and opportunities in the further improvement and applications. Emphasis especially focuses on the developments of mercury, bismuth, carbon and gold based electrode materials. It is expected that these novel electrochemical detection systems will succeed in on-site Cr(VI) measurements with excellent performance, reliable and convenient measurement, low cost and environmental effectiveness.
1. Introduction
Chromium (Cr) is an important chemical element in the periodic table with an atomic number of 24. The metal Cr in Cr(0) form is a steel-gray, hard and brittle metal with a high melting point and tarnish resistance, which is widely used for steel making.1 The two common oxidation states of Cr present in the environment, i.e. Cr(III) and Cr(VI), are extensively employed for chrome electroplating, dyes and pigments, leather tanning, and catalysis.2 Due to these industrial processes, large quantities of Cr compounds are discharged from liquid, solid, and gaseous waste streams into the environment, which can have substantially adverse biological and ecological effects.3,4Cr(III) and Cr(VI) have drastically different physicochemical properties and toxicity.5 Cr(III) is nearly insoluble at neutral pH and is considered to be a trace nutrient for the proper functioning of living organisms.6 It has been demonstrated to be responsible for the control of glucose and lipid metabolism in mammals. However, Cr(VI) compounds exert extremely hazardous effects on biochemical systems.7 It was found that Cr(VI) can easily penetrate the cell wall and then reduce to Cr(III) with the formation of reactive oxygen species (ROS), as shown in Fig. 1.8 The Cr(VI) itself and generated ROS can exert their oxidative potential and noxious influence towards the cell, leading to the inhibition of the metallo-enzyme system.9 At short-term exposure above the maximum contaminant level, Cr(VI) causes skin and stomach allergies or ulceration.10,11 Long-term exposure above maximum contaminant can cause damage to the liver, kidneys and nerve tissue, and even death in large doses.10,11 The United States Environmental Protection Agency (US EPA) has identified Cr(VI) as one of the 17 chemicals posing the greatest threat to humans.12 In order to protect human health and the environment, a maximum limit of 50 μg L−1 (1.0 μM) Cr(VI) in groundwater system was set by the World Health Organization (WHO).13 Furthermore, Cr(VI) compounds are significantly soluble and mobile in both biological and natural systems.12 Up to 220 μg L−1 (4.2 μM) naturally occurring Cr(VI) has been reported in the groundwater system of USA, while the Cr(VI) concentration in industrial waste generally varies from 40 to 1000 mg L−1 (0.8 to 19.2 μM).14 Therefore, selective detection and monitoring of Cr(VI) is significantly important in order to provide control of this highly toxic substance for human and environmental concerns.
 | ||
| Fig. 1 Schematic diagram of toxicity and mutagenicity of Cr(VI) (modified from ref. 8). | ||
Considerable investigations have been carried out to the quantitative techniques of Cr(VI) including spectroscopic, chromatographic and electrochemical approaches.15–18 However, the concentration of coexisting Cr(III) compounds are usually 10 to 1000 times higher than Cr(VI) concentrations in target analytes, resulting in serious interference for Cr(VI) quantification.19 Consequently, prior separation, reaction or complexing is necessary for many analytical methods, such as atomic absorption spectroscopy, chromatography, fluorescence, UV-vis spectroscopy and mass spectrometry, leading to the limitation of time-consuming procedures and the high cost of instruments.20 Recently, electrochemical detection of Cr(VI) has attracted increasing interest since it offers high sensitivity and powerful information, inherent miniaturization of both the detector and control system, good cost-effectiveness, minimal powder requirements, and excellent compatibility with advanced microfabrication technologies.21,22
This review discusses the recent advances in the electrode materials and detection strategies for electrochemical quantification of Cr(VI), compares the performances and properties of various detection systems, and outlines key challenges and opportunities in the further development and applications. Emphasis is mainly focused on the developments of mercury and bismuth based, carbons-based and gold-based electrode materials for Cr(VI) detection. Given the very broad field and long history of electrochemical Cr(VI) detection, this is not a comprehensive review but rather a view of recent important developments and applications, the authors apologize for the potential oversights of some important contributions.
2. Electroanalytical techniques
Electroanalytical methods are the interconversion between electricity and chemistry, which determine the electrical quantities of current, potential, or charge with respect to the change of chemical parameters.23 The type of electrical signal obtained for quantitation reflects the difference between many electroanalytical methods. These analytical techniques achieve a wide range of applications in food and environmental monitoring, industrial quality and safety control, or biomedical diagnoses.24There are two principal classes of electroanalytical techniques, i.e. potentiometric and potentiostatic.25–27 Potentiometry is a static (zero-current) method where the target analyte information is determined by the potential generate across an ion-selective membrane, while potentiostatic (controlled-potential) methods deal with the charge transfer (dynamic) processes at the electrode/solution interface.27 Potentiostatic methods can measure any chemical species including the electroactive compounds via reduction or oxidation, and the non-electroactive compounds via indirect or derivatization procedures. Compared to potentiometry, the advantages of potentiostatic methods are high sensitivity, selectivity for electroactive species, a wide linearity, portable and low-cost instrumentation and particularly a wide range of electrode materials availability, resulting in a significantly low detection limit even with very small (5–20 μL) sample volumes. Consequently, the reported Cr(VI) electrochemical detections are mainly focused on potentiostatic methods, including cyclic voltammetry (CV), chronoamperometry, differential pulse voltammetry (DPV) and stripping voltammetry. It has been demonstrated that different methods possess significantly distinct electrochemical detection performances as illustrated in Table 1.23
| Techniques | Working electrode | Detection limit (M) | Speed (min) | Response shape |
|---|---|---|---|---|
| a DC: direct current; AC: alternating current; DP: differential pulse; SW: square wave; DME: dropping mercury electrode; HDME: hanging dropping mercury electrode; MFE: mercury film electrode. | ||||
| DC polarography | DME | 10−5 | 3 | Wave |
| NP polarography | DME | 5 × 10−7 | 3 | Wave |
| DP polarography | DME | 10−8 | 3 | Peak |
| DP voltammetry | Solid | 5 × 10−7 | 3 | Peak |
| SW polarography | DME | 10−8 | 0.1 | Peak |
| AC polarography | DME | 5 × 10−7 | 1 | Peak |
| Chronoamperometry | Stationary | 10−5 | 0.1–2 | Peak |
| Cyclic voltammetry | Stationary | 10−5 | 0.1–2 | Peak |
| Stripping voltammetry | HDME, MFE | 10−10 | 3–6 | Peak |
| Adsorptive stripping voltammetry | HMDE | 10−10 | 2–5 | Peak |
| Adsorptive stripping voltammetry | Solid | 10−9 | 4–5 | Peak |
| Adsorptive catalytic stripping voltammetry | HMDE | 10−12 | 2–5 | Peak |
Besides, potentiostatic detection is determined by the electrochemical processes that occur at the electrode/solution interface, therefore the electrochemical cell at least requires the electrodes (conductors) and contacting sample solution (electrolyte).23 The electrode surface is a junction unit between an ionic conductor and an electronic conductor. Clearly, the electrode materials are of great importance for electrochemical detection, and there is a significant difference of detection performance between two electrodes even with the same electrochemical technique as shown in Table 1.23 Consequently, this review discusses the trend and developments of electrode materials and electrochemical techniques for electrochemical Cr(VI) detection with respect to the Cr(VI) detection performances such as detection limit, selectivity, linear range, response times and long-term stability.
3. Mercury and bismuth based electrodes
Compared to other analytical techniques for Cr(VI) quantification, electrochemical techniques appear significantly attractive because they allow direct redox speciation without any separation step such as chromatography, extraction, or ion-exchange resins.28 In particular, adsorptive stripping voltammetry with an effective preconcentration scheme offers a highly sensitive detection of trace Cr(VI) in connection to low-cost portable instrumentation.293.1 Mercury-based electrodes
In adsorptive stripping voltammetry, the target analyte is adsorbed on the working electrode during a preconcentration step and then oxidized/reduced from the electrode during the stripping process, while the current is measured during the stripping step.28 For the purpose of Cr(VI) detection, diethylenetriamine pentaacetic acid (DTPA), bipyridine, or cupferron is employed as a complexing agent.30 The hanging mercury drop electrode (HMDE) and mercury-film electrode (MFE) are generally employed in this type of detection, due to the fact that they can provide effective preconcentration, favorable redox reaction of the Cr(III) complex as well as a reproducible and renewable surface.31Boussemart et al.32 reported the determination of aqueous Cr(VI) ions using cathodic stripping voltammetry (CSV) via adsorptive collection of complex species with diethylenetriamine pentaacetic acid (DTPA) on a hanging mercury drop electrode in the year of 1992 as shown in Fig. 2. They identified that Cr(VI) as chromate is rapidly reduced to Cr(III) on the electrode surface and a Cr(III)–DTPA complex is generated, subsequently its reduction to a Cr(II)–DTPA species occurs with a well-defined reduction peak. The detection limit for chromium(VI) in distilled water is 10−11 M at a deposition time of 2 min, while the detection limit in sea water is 10−10 M; possibly due to major cation competition (of calcium and magnesium) for DTPA in the sea water. Cr(VI) produces a stable peak using the optimized CSV procedures, while the Cr(III) peak is unstable due to probable conversion of the chromium(III) complex to an electrochemically inert complex over a period of around 30 min. This different behavior of Cr(VI) and -(III) were used to determine reactive chromium(III) and chromium(VI), respectively.
 | ||
| Fig. 2 Cathodic stripping voltammograms of Cr(VI) in purified sea water (reproduced from ref. 32 with permission of Elsevier). | ||
In order to minimize the Cr(III) interference towards Cr(VI) detection, Grabarczyk and co-authors employed nitrilotriacetic acid (NTA) or ethylenediaminetetraacetic acid (EDTA) as a Cr(III) masking agent.33–35 This procedure allows for selective Cr(VI) detection in the presence of over thousand fold excess of Cr(III) or other common foreign ions. High selectivity and a wide concentration linear range (1 × 10−10 to 1 × 10−8 M) of Cr(VI) were obtained. This method was verified by analysis of Cr(VI) in natural water samples with satisfactory results.
However, the use of HMDE for Cr(VI) detection has some inconveniences such as the insufficient mechanical resistance and limited surface area of the mercury drop (≤3 mm2).36 Based on the adsorptive nature of Cr(III)–DTPA complex accumulation, the sensitivity and detection limit of Cr(VI) determination are greatly dependent on the surface area of the working electrode.37 Therefore, there is increasing interest in the mercury film electrode (MFE) for field measurements due to its virtue of robustness, mechanical stability, larger surface area and simple maintenance. Baś38 developed a refreshable mercury film silver based electrode [Hg(Ag)FE] where the thin liquid layer can be easily refreshed before each measurement. This type of film electrode is featured by its excellent surface reproducibility (not less than 2%) and long-term stability (1500–2000 cycles) with a mechanical refreshing time shorter than 1−2 s. Furthermore, the surface area of this electrode can be adjusted from 1.5 to 12 mm2, resulting in better sensitivity and detection limit of Hg(Ag)FE compared to HDME.
Based upon mechanical stability and simplicity of refreshed Hg(Ag)FE, Grabarczyk, Baś and Korolczuk39 applied this electrode for Cr(VI) detection in soil samples, making the field measurements possible. The DTPA is used as an extraction agent for the Cr(VI) in soil and then a complexing agent to form Cr(III)–DTPA for voltammetric determination, achieving the connection of extraction and following Cr(VI) detection in one cell, which significantly decreases the time of the whole measurement and eliminates the requirement for additional equipment and reagents.
3.2 Bismuth film electrodes
Recently, due to the toxicity, handling, volatility and disposal issues of mercury, novel alternative electrode materials with a similar performance are highly desirable to meet the increasing demands for on-site environmental monitoring of trace Cr(VI).40 In the last decade, bismuth film electrodes have appeared as a promising alternative.41 Compared to the common mercury electrodes, bismuth film electrodes offer well-defined and highly reproducible stripping responses, excellent resolution of neighboring peaks, high hydrogen evolution and wide linear range.42 Furthermore, bismuth is an environmentally-friendly element with very low toxicity, and its good mechanical stability enables operation under hydrodynamic conditions, which is particularly useful for filed measurements in flow systems.43Joseph Wang and co-authors44 developed a sensitive adsorptive stripping voltammetric system at a bismuth film coated glassy carbon electrode for trace measurements of chromium(VI) in the presence of diethylenetriamine pentaacetic acid (DTPA). Compared to the mercury electrode, the DTPA-based detection mechanism at the bismuth film electrode is similar, and the observed performance is also very comparable or even slightly better as illustrated in Fig. 3. Besides, the detection potential of −1.10 V at the Bi electrode is lower than the corresponding one of −1.22 V at the Hg electrode, suggesting a thermodynamic superiority. The attractive behavior of this novel “mercury-free” chromium sensor is promising for on-site environmental and industrial detection of chromium(VI).
 | ||
| Fig. 3 Stripping voltammograms of 20 nM Cr(VI) at mercury (A) and bismuth (B) coated glassy carbon electrodes. (Reproduced from ref. 44 with permission of Elsevier.) | ||
Furthermore, Jorge et al.45 explored an adsorptive stripping voltammetric protocol combined with a rotating-disc bismuth film electrode for the determination of chromium(VI) in the presence of DTPA. The rotating-disc electrode was employed to improve mass transport of chromium species to the electrode surface, therefore improved adsorptive accumulation process and sensitivity were obtained using this hydrodynamic configuration. Another interesting feature is the feasibility of this type of sensor to obtain simultaneous adsorptive stripping voltammetric detection of Cr(III) and Cr(VI) at trace levels, such as multivariate calibration methodology.
The substrate of bismuth film also plays an important role for the detection performance of Cr(VI). Compared to the traditional Bi film coated glassy carbon electrodes, Ouyang et al.46 developed an improved Bi film wrapped single walled carbon nanotubes modified glassy carbon electrode (Bi/SWNTs/GCE) as a highly sensitive protocol for ultratrace Cr(VI) detection. The introduction of negatively charged SWNTs significantly decreased the Bi particles size to nanoscale with more uniform and smoother morphology as illustrated in Fig. 4, improved the speed of electron transfer and hydrophilicity, facilitating the stripping voltammetric detection for Cr(VI) in aqueous system. A linear concentration range of 0–25 nM and a fairly low detection limit of 0.036 nM were obtained. Besides, this novel electrode exhibits better reproducibility and repeatability compared to Bi film modified GCE.
 | ||
| Fig. 4 Contact angles of bare electrode, and electrodes modified with Bi particles, SWNTs and Bi/SWNTs composite, respectively. (Reproduced from ref. 46 with permission of Elsevier.) | ||
In the real sample analysis, Cr(VI) is usually presented with other toxic heavy metals, such as Pb(II) and Cd(II).47 However, the electrochemical detection mechanism of Cr(VI) at bismuth film electrode is different from that used for Pb(II) and Cd(II). Anodic stripping voltammetry (ASV) is employed to detect Pb(II) and Cd(II) via their formation of alloys with bismuth, while the Cr(VI) determination is through its reduced form, i.e. Cr(III). In addition, the formation of the PbCrO4 complex between Pb(II) and Cr(VI) species makes the detection insensitive and inaccurate for both heavy metal ions. Therefore, in order to achieve the simultaneous detection of Pb(II), Cd(II) and Cr(VI), Erkang Wang and co-authors48 developed a combined H2O2-based reduction with stripping voltammetry method as presented in Fig. 5. By reducing the Cr(VI) to Cr(III) and releasing Pb(II) from PbCrO4, these three ions were successfully detected at the bismuth film electrode with a satisfactory detection limit. Besides, the detection approach was simplified and its sensitivity can be further improved by modifying the working electrode surface with a cation-exchange polymer, such as Nafion.
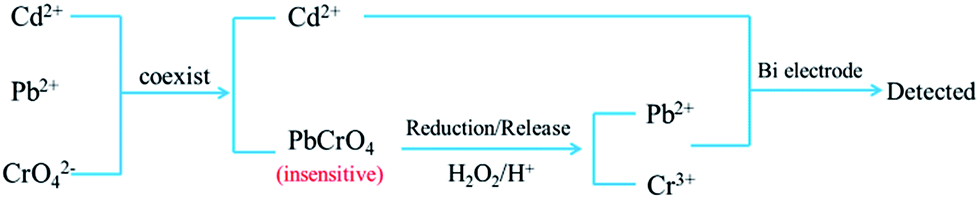 | ||
| Fig. 5 The schematic illustration of simultaneous detection of Cr(VI), Pb(II) and Cd(II) (modified from ref. 48). | ||
3.3 Comparison of the adsorptive stripping voltammetric determination for Cr(VI) at different systems
To better illustrate the effect of electrode materials, complexing/reduction agents and preconcentration, the Cr(VI) detection performances in different systems were compared in Table 2. Generally, the adsorptive stripping voltammetric detection of Cr(VI) at mercury or bismuth based electrodes has a preconcentration step and then the formed Cr(III) complex is detected with respect to the target Cr(VI) concentration.32 Therefore, a significantly low detection limit with concentration linear range was obtained, which is suitable for trace or ultra-trace Cr(VI) detection. However, the Cr(VI) concentration level in some groundwater systems in the USA is around 10 to 50 mg L−1 (0.2 to 1.0 × 10−6 M) or even as high as 220 μg L−1 (4.2 × 10−6 M), while the data in industrial wastewater range from 40 to 1000 mg L−1 (0.8 to 19.2 × 10−6 M). Clearly, the linear concentration range does not meet the requirement of field measurement in many cases, which is possibly due to the small electrode surface area. Besides, this is an indirect detection of Cr(VI) and the produced Cr(III) complex is actually detected at the electrode, resulting in Cr(III) interference or relating complicated and time-consuming Cr(III) masking procedures. Consequently, an electrochemical method for the facile and direct detection of Cr(VI) is highly desirable.| Electrode | Agent | Accumulation (minutes) | Linear range (M) | Detection limit (M) | Sample solution | Ref. |
|---|---|---|---|---|---|---|
| a HDME: hanging dropping mercury electrode; DTPA: diethylenetriamine pentaacetic acid. | ||||||
| HDME | DTPA | 2 | — | 10−10 | Sea water (pH 5.2) | 32 |
| 2 | — | 10−11 | Buffer (pH 6.4) | |||
| HDME | Bipyridine | 1 | 5 × 10−10 to 5 × 10−9 | 1.4 × 10−10 | Buffer (pH 8.0) | 75 |
| HDME | DTPA | 0.5 | 1 × 10−9 to 1 × 10−7 | 3.8 × 10−10 | Buffer (pH 6.1) | 30 |
| Hg(Ag) film | DTPA | 0.5 | 2 × 10−10 to 2.2 × 10−9 | 5 × 10−11 | Buffer (pH 6.2) | 76 |
| Hg(Ag) film | DTPA | 0.33 | 5 × 10−10 to 5 × 10−8 | 1.9 × 10−10 | Buffer (pH 6.2) | 38 |
| Hg(Ag) film | DTPA | 0.17 | 10−9 to 4 × 10−8 | 2.6 × 10−10 | Extracted solution | 39 |
| From solid (pH 6.2) | ||||||
| Bi film-GC | DTPA | 2 | 5 × 10−9 to 5 × 10−8 | 3 × 10−10 | Buffer (pH 6.0) | 44 |
| Bi film/GC RDE | DTPA | 2 | — | 3.3 × 10−10 | Buffer (pH 6.0) | 45 |
| Bi film-GC | H2O2 | 2 | 2 × 10−10 to 1.3 × 10−9 | 1 × 10−10 | Buffer (pH 4.5) | 48 |
| Bi/SWNTs/GC | DTPA | 2.3 | 10−9 to 2.5 × 10−8 | 3.3 × 10−10 | Buffer (pH 4.5) | 46 |
4. Carbon-based electrodes
Carbon materials possess unique properties such as high electrical conductivity, chemical and mechanical stability. Besides, their morphologies enable their functionality and stable operation in the design of electrochemical processes. Therefore, they are widely incorporated as sensing elements to obtain higher sensitivity and lower detection limit.494.1 Carbon paste electrodes
Cr(VI) detection with adsorptive stripping voltammetry often couples with reactions involving suitable complexing agents, such as DTPA or 2,2′-bipyridine, to obtain a Cr(III) complex which is detected at the electrode surface.32,39 Recently, the detection procedures were significantly simplified at carbon paste electrodes (CPEs). The complexing agents (quarternary ammonium salts) was admixed as modifiers of electrode to efficiently accumulate the Cr(VI) ion-pairs, and then the Cr(VI) was detected via its direct cathodic reduction as shown in Fig. 6.50 The signal of interest was reproducible within ±8% and linear to the concentration in a range of 0.5–50 × 10−6 M CrO42−, with a detection limit of about 5 × 10−8 M (with accumulation for 300 s). Furthermore, electrochemical sensors based on carbon paste electrodes are economic and easy to construct, and have good reproducibility and excellent functional lifetime. | ||
| Fig. 6 Stripping voltammograms of Cr(VI) at the micromolar concentration level. (1) Blank; (2–4) 1, 5, 10 μM Cr(VI). (Reproduced from ref. 50 with permission of Elsevier.) | ||
Potentiometry is another important analytical technique due to its simplicity, low cost, rapid response, selectivity as well as ability to test the analytes in colored and turbid samples.51 Electrodes of solid-state membranes based upon carbon paste and composite platforms are considered to be an economic and robust potentiometric, which present stable electrochemical responses, lower ohmic resistance and better functional lifetime compared to polymeric membranes based potentiometric sensor.51,52 Sanchez-Moreno developed a diphenylcarbazide modified carbon paste electrode for the selective and direct detection for Cr(VI) ions.53 A significant wide linear response range from 1.00 × 10−6 to 1.00 × 10−2 M and a low detection limits of 9 × 10−7 M were obtained. This sensor also exhibited high selectivity to Cr(VI) even in the presence of Cr(III) or other common ions present in industrial or environmental samples.
4.2 Modified glassy carbon electrode
Glassy carbon is an excellent electrode substrate (current collector) material for constructing the sensing protocol due to its high density and low porosity. After polishing, a smooth surface can be obtained to minimize the substrate double layer current and provide good affinity for noble metal deposition or surface modification.54 Carrington et al. reported a pyridine-functionalized sol–gel film coated glassy carbon electrode for Cr(VI) detection using square-wave voltammetry.21 The interaction between the pyridinium groups in the sol–gel film and Cr(VI) anions in solution results in a wide linear range for Cr(VI) analysis from 11.7 to 400 ppb and low detection limit (4.6 ppb). Besides, the modified electrode exhibited excellent stability, reproducibility and good resistance to other metal ions (even 105 excess Cr(III)).| CrO42− + 8H+ → Cr3+ + 4H2O − 3e− | (1) |
Prussian blue (iron(III) hexacyanoferrate) is another well-known bimetallic mixed valence inorganic compound, which can be employed as a modifier of the glassy carbon-based electrode for many electroanalytical applications such as arsenite and H2O2 detection.55,56 Xing et al.57 developed a simple and disposable amperometric detection of trace Cr(VI) using Prussian blue modified glassy carbon electrode (PB/GCE). The Prussian blue PB film was identified to mediate the Cr(VI) reduction, and the resulting PB/GCE provided a wide linear range for Cr(VI) detection (0.5 to 200 ppb) and low detection limit (0.15 ppb). In addition, the as-prepared electrode was successfully applied to trace Cr(VI) determination in wastewater, exhibiting excellent stability and resistance to other metal ions or surfactants.
4.3 Carbon nanotubes
Since the discovery of arc-grown carbon nanotubes, there has been considerable theoretical and practical investigation to their properties. The carbon nanotubes (CNTs) possess superiority such as large surface area, excellent electron transfer ability and easy surface-modification, resulting in their wide application as an electrochemical sensor.58 Rudnitskaya et al.59 fabricated a conductive lignin–poly(propylene oxide) copolymer doped by carbon nanotubes as a potentiometric sensor for Cr(VI). The lignin/CNTs based sensors presented a very low or no response to all alkali, alkali-earth and transition metal cations except Cr(VI) at pH 2. More importantly, a wide linear range from 10−5 M to 10−2 M and a low detection limit of 5 × 10−6 M were obtained, suggesting it as a promising material for Cr(VI)-sensitive potentiometric sensors.Surface functionalization of the CNTs can also help in endowing the electrocatalytic reduction activity and corresponding detection for heavy metals. Deep et al.60 fabricated phosphinic acid derivative functionalized single-walled carbon nanotubes (SWCNTs) for the ultra-trace Cr(VI) detection. A linear response over a range of Cr(VI) concentrations (0.01–10 ppb) and a detection limit of 0.01 ppb were obtained using amperometry. The practical utility of the proposed sensor is demonstrated by determining the Cr(VI) concentration in an industrial effluent sample and an underground water sample.
4.4 Screen-printed carbon electrode
Screen-printed carbon electrodes (SPCEs) are a highly attractive alternative for Cr(VI) detection due to their relatively low costs and size reduction, chemical stability, wide working potential window, rich surface chemistry of carbon materials employed allowing various chemical derivatization or modification.61 Hallam et al.62 reported graphite screen printed macroelectrodes for the electroanalytical sensing of low ppb levels of Cr(VI) in aqueous solutions. There is a good linear relationship for Cr(VI) determination in aqueous solutions over the range 100 to 1000 μg L−1 with a limit of detection of 19 μg L−1, and this analytical protocol is presented to be applicable for Cr(VI) sensing in real water samples at levels set by the World Health Organisation.Furthermore, the development of SPCE has led to the assembly of an intelligent sensors platform which can be integrated into portable systems.63 Miscoria et al.64 developed SPE graphite electrodes by integrating working and counter electrode into a unique strip as presented in Fig. 7. Bergamini et al.22 reported the fabrication of poly-L-histidine film modified SPCE and its integration with reference and the auxiliary electrodes onto an alumina ceramic base, which was employed as a convenient Cr(VI) sensor. These sensing platforms exhibited good sensitivities and reproducibility, an extended dynamic range, and a low detection limit. In addition, the sensor presented great resistance towards Cr(III) interference.
 | ||
| Fig. 7 (A) Layout of SPEs, single bar and as complete substrate for batch printing on alumina (inset). (B) Ink deposition steps. (Reproduced from ref. 64 with permission of Elsevier.) | ||
4.5 Comparison of Cr(VI) detection at carbon-based electrodes
The comparison of Cr(VI) detection performance at different carbon-based electrodes is presented in Table 3. Compared to the Hg and Bi based electrodes, the detection target at carbon-based electrodes is Cr(VI) itself, resulting in improved selectivity and simplified procedures. Besides, the linear concentration range at carbon electrodes is wider, which can reach 10−2 M or even 10−1 M. However, the detection limit of some carbon-based protocols is not comparable to the corresponding ones at Hg and Bi based electrode, but they still meet the detection requirement of WHO at 10−6 M Cr(VI).| Electrode | Method | Accumulation (minutes) | Linear range (M) | Detection limit (M) | Sample solution | Ref. |
|---|---|---|---|---|---|---|
| a SPEs: screen printed electrodes; MWCNTs: multi-wall carbon nanotubes; SWCNTs: single-wall carbon nanotubes. | ||||||
| Modified carbon paste | Stripping voltammetry | 5 | 5 × 10−7 to 5 × 10−5 | 10−8 to 8 × 10−8 | 0.3 M HCl + 0.1 M NaCl | 50 |
| Graphite epoxy | Potentiometry | 0.3 | 1 × 10−6 to 1 × 10−2 | 6.3 × 10−7 | pH 3.0 | 53 |
| Carbon paste | Potentiometry | 0.4 | 1 × 10−6 to 1 × 10−2 | 9 × 10−7 | pH 3.0 | 53 |
| Polypyrrole graphite | Potentiometry | 1.3 | 1 × 10−6 to 1 × 10−1 | 5 × 10−7 | pH 7.0 | 77 |
| Lignin–poly(propylene oxide) doped MWCNTs | Potentiometry | — | 1 × 10−5 to 1 × 10−2 | 5 × 10−6 | pH 7.0 | 59 |
| Graphite expoxy–SPEs | Potentiometry | 0.3 | 10−6 to 3.2 × 10−4 | 7.7 × 10−7 | pH 3 | 78 |
| Sol–gel film modified glassy carbon | Square-wave voltammetry | 10 | 2 × 10−10 to 7.7 × 10−9 | 9 × 10−11 | 0.1 M HCl | 21 |
| Poly-L-histidine/SPEs | Linear sweep voltammetry | 3 | 10−7 to 1.5 × 10−4 | 4.6 × 10−8 | pH 4 | 22 |
| Graphite–SPEs | Linear sweep voltammetry | No | 2 × 10−6 to 2 × 10−5 | 3.6 × 10−7 | pH 1 | 64 |
| Graphite–SPEs | Amperometry | No | 3 × 10−6 to 10−2 | 10−6 | pH 1 | 64 |
| Glucose oxidase–SPEs | Amperometry | No | 9 × 10−8 to 7.7 × 10−7 | 9 × 10−8 | pH 4 | 79 |
| Prussian blue modified glassy carbon | Amperometry | No | 10−11 to 3.8 × 10−9 | 2.9 × 10−12 | 0.1 M KCl + 0.1 M HCl | 57 |
| Phosphinic modified SWCNTs | Amperometry | No | 2 × 10−13 to 2 × 10−10 | 2 × 10−13 | 0.1 M H2SO4 | 60 |
Different functionalization offers significant improvement for Cr(VI) detection at carbon electrodes, direct and convenient measurement is achieved using potentiostatic or potentiometric techniques. Furthermore, the integrated sensor appears to be a promising sensitive, facile and reliable detection protocol for on-site Cr(VI) determination for environmental and industrial monitoring.
5. Gold-based electrodes
Another efficient electrocatalyst for Cr(VI) reduction and corresponding detection is gold materials. Burke et al. compared the Cr(VI) electrochemical reduction behavior between platinum and gold electrodes.65 It was found the reduction reaction occurred rapidly at a much lower over-potential on gold with respect to platinum. Welch and Compton identified that Cr(VI) can be directly detected at a polycrystalline Au electrode by cyclic voltammetry.13 A detection limit of 4.3 μM Cr(VI) in the presence of 5 mM Cr(III), and a Cr(VI) linear concentration range of 100–1500 μM were obtained. Besides, this electrochemical detection also exhibited good interferences towards common environmental species, such as Ni2+, Cu2+, Fe3+, Cr3+ and surfactants.5.1 Gold modified carbon electrodes
Based upon the investigation on polycrystalline gold electrode, considerable efforts have been carried out on modifying the electrode materials to improve the Cr(VI) detection performance. Kachoosangi and Compton66 reported linear sweep voltammetric determination of chromium(VI) at a gold plated carbon composite electrode in water; the bare composite electrode exhibited no sensitivity for Cr(VI) reduction even for a high concentration of ∼50 μM, while the gold plated electrode presented a high sensitivity for Cr(VI) reduction and obtained a well-defined peak as shown in Fig. 8. Therefore, a wide linear range between 20 and 2000 μg L−1 and a detection limit of 4.4 μg L−1 Cr(VI) were obtained. This low cost, simple and direct method achieves a wide range of calibration curves, suggesting that this proposed approach can be easily employed for routine analysis of Cr(VI) in severely polluted water samples in the lab and field.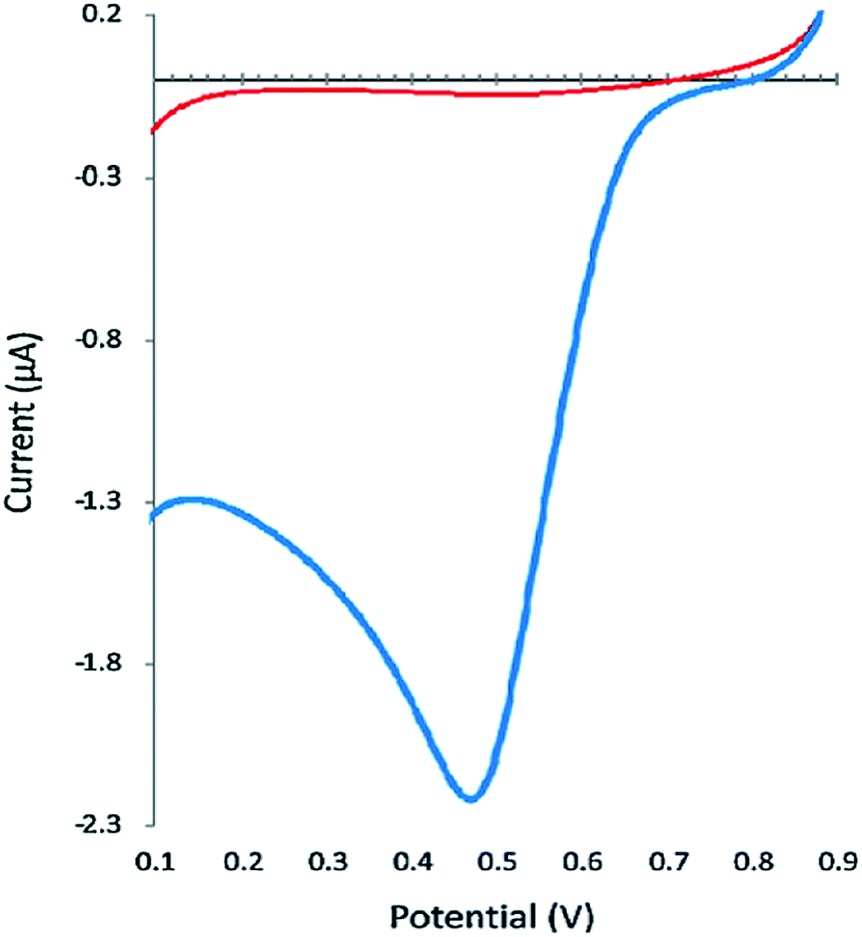 | ||
| Fig. 8 The linear sweep voltammetric reduction of 2600 μg L−1 Cr(VI) on bare composite electrode (straight line) versus gold plated carbon composite electrode (sharp peak) in 0.3 M nitric acid, with a scan rate of 50 mV s. (Reproduced from ref. 66 with permission of Elsevier.) | ||
Another example of gold modified electrodes for Cr(VI) detection is gold screen printed macroelectrodes (AuSPEs). Compared to polycrystalline gold macroelectrodes, AuSPEs have comparable analytical performance towards Cr(VI), while possessing additional advantages due to their disposable one-shot nature, the ease of mass production, and no need to potential cycle for the required gold oxide formation.67 Consequently, the analytical protocol of Cr(VI) at AuSPEs is significantly simplified. Banks et al.68 developed such a detection method to achieve Cr(VI) detection in aqueous solutions over the range 10 to 1600 mM with a detection limit of 4.4 mM. The feasibility of this method was also tested through Cr(VI) detection in environmental samples.
5.2 Gold nanoparticles modified electrodes
Recently, owing to the attractive physicochemical and electrocatalytical properties of nanometric noble metal particles, gold nanoparticles modified electrodes have been widely developed for Cr(VI) detection.69 Jin et al.70 reported a simple and effective electrochemical Cr(VI) detection using the Au nanoparticle modified titania nanotubes (TiO2 NTs) electrode as shown in Fig. 9. It was identified that the electrochemical reduction of Cr(VI) at the Ti/TiO2 NT/Au electrode presented an around 23 fold enhancement in activity with respect to the polycrystalline gold electrode, due to the superior conduction capability and high surface-to-volume ratio of the nanoparticle/nanotubular structure. Therefore, the Ti/TiO2 NT/Au electrode exhibited a wide linear concentration range from 0.10 mM to 105 mM, a low detection limit of 0.03 mM, and a high sensitivity of 6.91 mA mM−1 for Cr(VI) determination via chronoamperometry, satisfying the detection standards of the World Health Organization. Besides, this electrode shows good resistance against the interference from coexisting Cr(III) and other metal ions, and excellent Cr(VI) recovery from both tap and lake water samples. | ||
| Fig. 9 (Upper) SEM image of the as prepared Au nanoparticle decorated TiO2 nanotube electrode; (lower) amperometric current responses as the result of the successive addition of Cr(VI) at the electrode potential of 0.28 V in a 0.1 M HCl solution. The inset is the enlarged amperometric responses of the low Cr(VI) concentration area. (Reproduced from ref. 70 with permission of The Royal Society of Chemistry.) | ||
In order to improve the Cr(VI) detection performance, considerable efforts have been devoted to the functionalization and modification of the nanoparticle itself and/or its support materials. Ouyang et al.71 reported the fabrication of a flower-like self-assembly of gold nanoparticles (Au NPs) supported glassy carbon electrode as a highly sensitive protocol for ultratrace Cr(VI) detection. After functionalization by a thiol pyridinium, the as-prepared electrode presented a well-defined peak for selective Cr(VI) reduction with a linear concentration range of 10–1200 ng L−1 and a low detection limit of 2.9 ng L−1. Santhosh72 reported a highly sensitive and selective Cr(VI) sensor using Au nanoparticles (Au NPs) decorated graphene nanosheet matrix. Such a nanocomposite film combines the advantages of Au NPs and graphene due to the excellent synergistic effect, facilitating the electron-transfer processes and leading to 100 times Cr(VI) reduction activity compared to the polycrystalline gold electrode. Further systematic investigation about the Cr(VI) detection at gold nanoparticles supported graphene electrodes using other electrochemical techniques or integrated sensor is particularly encouraged.
5.3 Comparison of Cr(VI) detection at gold based electrode
As illustrated in Table 4, there is good electrocatalytic activity for Cr(VI) reduction at macro gold or related electrode. However, the detection limit at these electrodes does not satisfy the WHO requirement (1.0 μM Cr(VI)). In order to improve the detection performance, the gold nanoparticles modified electrodes were utilized due to their attractive physicochemical and electrocatalytic properties. Therefore, significant enhancement of the linear range and detection limit for Cr(VI) were obtained in gold nanoparticles modified electrode via direct and facile electrochemical techniques, and it should be noted that the as-obtained performance is even comparable with the ones at Hg and Bi film electrodes via relatively complicated preconcentration and stripping methods.| Electrode | Method | Linear range (M) | Detection limit (M) | Sample solution | Ref. |
|---|---|---|---|---|---|
| a NPs: nanoparticles. | |||||
| Au NPs on glass carbon electrode | Stripping square voltammetry | 2 × 10−10 to 2.3 × 10−8 | 5.6 × 10−11 | pH 4.5 | 71 |
| Azacrown decorated Au | Electrochemical impedance | 2 × 10−11 to 2 × 10−9 | 2.7 × 10−14 | pH 5.0 | 80 |
| Au NPs on carbon screen printed | Differential pulse voltammetry | 4 × 10−7 to 3 × 10−5 | 4 × 10−7 | pH 6.0 | 81 |
| Polycrystalline Au | Cyclic voltammetry | 10−4 to 1.5 × 10−3 | 4.3 × 10−6 | 0.1 M HCl | 13 |
| Au NPs on indium tin oxide | Cyclic voltammetry | 5 × 10−6 to 10−4 | 2 × 10−6 | 0.01 M NaCl + 0.01 M HCl | 82 |
| Au screen printed macro electrode | Linear sweep voltammetry | 10−5 to 1.6 × 10−3 | 4.4 × 10−6 | 0.05 M H2SO4 | 67 |
| Au microelectrode | Linear sweep voltammetry | 10−7 to 3.8 × 10−6 | 10−9 | 0.03 M HNO3 | 83 |
| Au film modified carbon composite | Linear sweep voltammetry | 3.8 × 10−7 to 3.8 × 10−5 | 8.4 × 10−8 | 0.3 M HNO3 | 66 |
| Au NPs on TiO2 nanotubes | Amperometry | 10−7 to 10−4 | 3 × 10−8 | 0.1 M HCl | 70 |
| Au NPs on indium tin oxide | Amperometry | 5 × 10−7 to 5 × 10−5 | 10−7 | 0.02 M NaCl + 0.01 M HCl | 82 |
| Au NPs on silicate network | Amperometry | 4 × 10−12 to 5.7 × 10−11 | 2 × 10−12 | 0.1 M HCl | 84 |
Significantly different detection behaviors for Cr(VI) determination were obtained dependent on the electrode materials system, including gold catalyst itself and also the catalyst support. Besides, the amperometric method possessed a lower detection limit than its linear sweep voltammetric detection at Au NPs on indium tin oxide electrode, suggesting electrochemical methods play an important role in the Cr(VI) detection performance.
6. Conclusions and prospects
This review has summarized recent advances in electrochemical detection of toxic Cr(VI) with respect to different electrochemical techniques and electrode materials. Compared to previous spectroscopic and chromatographic quantification approaches for Cr(VI), the adsorptive stripping voltammetric detection based on mercury and bismuth electrodes offers a highly sensitive Cr(VI) detection approach without any pre-separation step via the reduced Cr(III)-complex species. In order to simplify the detection step, improve the selectivity and increase the linear range, cost-effective carbon-based electrodes were developed to determine the Cr(VI) via its direct reduction behaviour. Recently, due to the attractive properties of gold nanoparticles, there is increasing interest in novel gold-based electrodes for the direct detection of Cr(VI) with excellent sensitivity, wide linear range, facile preparation and operation. Most of the Cr(VI) detection systems satisfy the detection requirement of the WHO. Therefore, coupling with the modern integrated micro-fabrication technology, these novel electrochemical Cr(VI) detection systems are expected to succeed in the on-site Cr(VI) measurements with excellent performance, reliable and convenient measurement, cost and environmental effectiveness.It should be noted that there are still many challenges and opportunities for the next-generation electrochemical Cr(VI) sensors: (1) most of the recently available electrochemical Cr(VI) detection focuses on the media of acidic aqueous solution or below the pH of 7, and scarce investigation has been carried out in alkaline solution possibly due to the low electrocatalytic activity for Cr(VI) reduction in this region.73,74 Therefore, it is highly desirable to develop novel electrodes which are capable of detecting Cr(VI) with comparable performance in alkaline solution. (2) In order to facilitate the on-site and in site electrochemical detection of Cr(VI), facile and reliable sampling methods in complicated systems, such as the soil and human body, are particularly required. (3) The significant bottleneck between laboratory research and widespread practical applications is needed to be bridged. The commercial sensor arrays based on the fundamental investigation are encouraged to be produced and applied in the real samples testing.
Due to the significant importance in human and environmental safety, electrochemical Cr(VI) detection will continue attracting more and more interest from the academic and practical aspects.
Notes and references
- J. W. Ball and D. K. Nordstrom, J. Chem. Eng. Data, 1998, 43, 895–918 CrossRef CAS.
- W. Jin, S. Zheng, H. Du, H. Xu and Y. Zhang, Ind. Eng. Chem. Res., 2010, 49, 8244–8247 CrossRef CAS.
- C. S. Gad, Sci. Total Environ., 1989, 86, 149–157 CrossRef.
- W. Jin, Z. Zhang, G. Wu, R. Tolba and A. Chen, RSC Adv., 2014, 4, 27843–27849 RSC.
- L. E. Eary and D. Rai, Environ. Sci. Technol., 1987, 21, 1187–1193 CrossRef.
- D. Rai, B. M. Sass and D. A. Moore, Inorg. Chem., 1987, 26, 345–349 CrossRef CAS.
- M. Cespon-Romero, M. C. Yubru-Biurru and M. P. Bermejo-Barrera, Anal. Chim. Acta, 1996, 327, 37–45 CrossRef.
- J. B. Vincent, Chromium: biological relevance, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, 2nd edn, Wiley, New York, 1994 Search PubMed.
- M. Costa, Toxicol. Appl. Pharmacol., 2003, 188, 1–5 CrossRef CAS.
- M. Pesti, Z. Gazdag and J. Belagyi, FEMS Microbiol. Lett., 2000, 182, 375–380 CrossRef CAS PubMed.
- C. Cervantes, J. Campos-Garcia, S. Devars, G. Gutierrez-Corona, H. Loza-Tavera, J. C. Torres-Guzman and R. Moreno-Sanchez, FEMS Microbiol. Rev., 2001, 25, 335–347 CrossRef CAS PubMed.
- J. Kotas and Z. Stasicka, Environ. Pollut., 2000, 107, 263–283 CrossRef CAS.
- C. M. Welch, O. Nekrassova and R. G. Compton, Talanta, 2005, 65, 74–80 CAS.
- State of the Science of Hexavalent Chromium in Drinking Water, Water Research Foundation, 2012.
- A. G. Cox, I. G. Cook and C. W. McLeod, Analyst, 1985, 110, 331–333 RSC.
- G. Y. Jung, Y. S. Kim and H. B. Lim, Anal. Sci., 1997, 13, 463–467 CrossRef CAS.
- A. Krushevska, A. Waheed, J. Nobrega, D. Amarisiriwardena and R. M. Barnes, Appl. Spectrosc., 1998, 52, 205–211 CrossRef CAS.
- R. Millacic and J. Stupar, Analyst, 1994, 119, 627–632 RSC.
- K. Ndungu, N.-K. Djane, F. Malcus and L. Mathiasson, Analyst, 1999, 124, 1367–1372 RSC.
- Y. Li and H. Xue, Anal. Chim. Acta, 2001, 448, 121–134 CrossRef CAS.
- N. A. Carrington, L. Yong and Z.-L. Xue, Anal. Chim. Acta, 2006, 572, 17–24 CrossRef CAS PubMed.
- M. F. Bergamini, D. P. dos Santos and M. V. B. Zononi, Sens. Actuators, B, 2007, 123, 902 CrossRef CAS PubMed.
- J. Wang, Analyticl Electrochemistry, John Wiely& Sons, 2nd edn, 2006 Search PubMed.
- J. Wang, Talanta, 2002, 56, 223–231 CrossRef CAS.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solutions, IUPAC, New York, 1985 Search PubMed.
- G. Korotcenkov, S. D. Han and J. R. Stetter, Chem. Rev., 2009, 109, 1402–1433 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, 2001 Search PubMed.
- J. Wang, Electroanalysis, 2005, 17, 15–16 CrossRef PubMed.
- A. Economou and P. R. Fielden, Analyst, 2003, 128, 205–213 CAS.
- M. Grabarczyk, Electroanalysis, 2008, 20, 2217–2222 CrossRef CAS PubMed.
- Z. Gao and K. S. Siow, Electroanalysis, 1996, 8, 602–606 CrossRef CAS PubMed.
- M. Boussemart, C. M. G. van den Berg and M. Ghaddaf, Anal. Chim. Acta, 1992, 262, 103–115 CrossRef CAS.
- M. Grabarczyk and M. Korolczuk, Anal. Bioanal. Chem., 2003, 376, 1115–1118 CrossRef CAS PubMed.
- M. Grabarczyk, L. Kaczmarek and M. Korolczuk, Electroanalysis, 2004, 16, 1503–1507 CrossRef CAS PubMed.
- M. Grabarczyk, L. Kaczmarek and M. Korolczuk, Anal. Bioanal. Chem., 2008, 390, 979–986 CrossRef CAS PubMed.
- J. A. Cox and P. J. Kulesza, Anal. Chim. Acta, 1983, 154, 71–78 CrossRef CAS.
- B. Baś and Z. Kowalski, Electroanalysis, 2002, 14, 15–16 CrossRef.
- B. Baś, Anal. Chim. Acta, 2006, 570, 195–201 CrossRef PubMed.
- M. Grabarczyk, B. Baś and M. Korolczuk, Microchim. Acta, 2009, 164, 465–470 CrossRef CAS.
- J. Wang, J. Lu, S. B. Hocevar, P. A. M. Farias and B. Ogorevc, Anal. Chem., 2000, 72, 3218–3222 CrossRef CAS.
- S. B. Hocevar, B. Ogorevc, J. Wang and B. Pihlar, Electroanalysis, 2002, 14, 1707–1712 CrossRef CAS PubMed.
- V. Rehacek, I. Hotovy, M. Vojs, T. Kups and L. Spiess, Electrochim. Acta, 2012, 63, 192–196 CrossRef CAS PubMed.
- T. Romann and E. Lust, Electrochim. Acta, 2010, 55, 5746–5752 CrossRef CAS PubMed.
- L. Lin, N. S. Lawrence, S. Thongngamdee, J. Wang and Y. Lin, Talanta, 2005, 65, 144–148 CrossRef CAS PubMed.
- E. O. Jorge, M. M. Rocha, I. T. E. Fonseca and M. M. M. Neto, Talanta, 2010, 81, 556–564 CrossRef CAS PubMed.
- R. Ouyang, W. Zhang, S. Zhou, Z.-L. Xue, L. Xu, Y. Gu and Y. Miao, Electrochim. Acta, 2013, 113, 686–693 CrossRef CAS PubMed.
- C. T. Dillon, P. A. Lay, A. M. Bonin, N. E. Dixon, T. J. Collins and K. Kostka, Carcinogenesis, 1993, 14, 1875–1880 CrossRef CAS PubMed.
- J. Li, J. Zhang, H. Wei and E. Wang, Analyst, 2009, 134, 273–277 RSC.
- M. N. Abbas and G. A. E. Mostafa, Anal. Chim. Acta, 2003, 478, 329–335 CrossRef CAS.
- I. Svancara, P. Foret and K. Vytras, Talanta, 2004, 64, 844–852 CrossRef CAS PubMed.
- M. J. Gismera, J. R. Procopio, M. T. Sevilla and L. Hernandez, Electroanalysis, 2003, 15, 126–132 CrossRef CAS PubMed.
- E. Khaled, H. N. A. Hassan, A. Girgis and R. Metelka, Talanta, 2008, 77, 737–743 CrossRef CAS PubMed.
- R. A. Sanchez-Moreno, M. J. Gismera, M. T. Sevilla and J. R. Procopio, Sens. Actuators, B, 2010, 143, 716–723 CrossRef CAS PubMed.
- J. Wang, M. Li, Z. Shi, N. Li and Z. Gu, Anal. Chem., 2002, 74, 1993–1997 CrossRef CAS.
- D. Moscone, D. D. Ottavi and D. Compagnone, Anal. Chem., 2001, 73, 2529–2535 CrossRef CAS.
- D. Zhang, K. Zhang, Y. L. Yao, X. H. Xia and H. Y. Chen, Langmuir, 2004, 20, 7303–7307 CrossRef CAS PubMed.
- S. Xing, H. Xu, G. Shi, J. Chen, L. Zeng and L. Jin, Electroanalysis, 2009, 21, 1678–1684 CrossRef CAS PubMed.
- L. Wang, X. Wang, G. Shi, C. Peng and Y. Ding, Anal. Chem., 2012, 84, 10560–10567 CrossRef CAS PubMed.
- A. Rudnitskaya, D. V. Evtuguin, L. C. Costa, M. P. F. Graca, A. J. S. Fernandes, M. R. P. Correia, M. T. S. R. Gomes and J. A. B. P. Oliveira, Analyst, 2013, 138, 501–508 RSC.
- A. Deep, A. L. Sharma, S. K. Tuteja and A. K. Paul, J. Hazard. Mater., 2014, 278, 559–565 CrossRef CAS PubMed.
- M. Tudorache and C. Bala, Anal. Bioanal. Chem., 2007, 388, 565–578 CrossRef CAS PubMed.
- P. M. Hallam, D. K. Kampouris, R. O. Kadara and C. E. Banks, Analyst, 2010, 135, 1945–1952 RSC.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Electrochem. Commun., 2009, 11, 1377–1380 CrossRef CAS PubMed.
- S. A. Miscoria, C. Jacq, T. Maeder and R. M. Negri, Sens. Actuators, B, 2014, 195, 294–302 CrossRef CAS PubMed.
- L. D. Burke and P. F. Nugent, Electrochim. Acta, 1997, 42, 399–411 CrossRef CAS.
- R. T. Kachoosangi and R. G. Compton, Sens. Actuators, B, 2013, 178, 555–562 CrossRef CAS PubMed.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2011, 136, 1067–1076 RSC.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2012, 137, 896–902 RSC.
- K. Sohn, F. Kim, K. C. Pradel, J. Wu, Y. Peng, F. Zhou and J. Huang, ACS Nano, 2009, 3, 2191–2198 CrossRef CAS PubMed.
- W. Jin, G. Wu and A. Chen, Analyst, 2014, 139, 235–241 RSC.
- R. Ouyang, S. A. Bragg, J. Q. Chamers and Z.-L. Xue, Anal. Chim. Acta, 2012, 722, 1–7 CrossRef CAS PubMed.
- C. Santhosh, M. Saranya, R. Ramachandran, S. Felix, V. Velmurugan and A. N. Grace, J. Nanotechnol., 2014, 1–7 CrossRef PubMed.
- W. Jin, M. S. Moats, S. Zheng, H. Du, Y. Zhang and J. D. Miller, Electrochim. Acta, 2011, 24, 8311–8318 CrossRef PubMed.
- W. Jin, M. S. Moats, S. Zheng, H. Du, Y. Zhang and J. D. Miller, J. Phys. Chem. B, 2012, 116, 7531–7537 CrossRef CAS PubMed.
- M. Korolczuk, Electroanalysis, 1999, 11, 1218–1221 CrossRef CAS.
- B. Baś, A. Bugajna, M. Jakubowska and E. Niewiara, Electroanalysis, 2012, 24, 2157–2164 CrossRef PubMed.
- R. Ansari, A. F. Delavar and A. Mohammad-khah, Microchim. Acta, 2012, 178, 71–79 CrossRef CAS.
- R. A. Sanchez-Moreno, M. J. Gismera, M. T. Sevilla and J. R. Procopio, Electroanalysis, 2011, 23, 287–294 CrossRef CAS PubMed.
- A. Calvo-Perez, O. Dominguez-Renedo and M. Alonso-Lomillo, Anal. Chim. Acta, 2014, 833, 15–21 CrossRef CAS PubMed.
- J. Wei, Z. Guo, X. Chen, D.-D. Han, X.-K. Wang and X.-J. Huang, Anal. Chem., 2015, 87(3), 1991–1998 CrossRef CAS PubMed.
- O. Dominguez-Renedo, L. Ruiz-Espelt, N. Garcia-Astrogano and M. J. Arcos-Martinez, Talanta, 2008, 76, 854–858 CrossRef CAS PubMed.
- M.-C. Tsai and P.-Y. Chen, Talanta, 2008, 76, 533–539 CrossRef CAS PubMed.
- E. A. Zakharova, E. E. Elesova, A. A. Skorokhodova and G. N. Noskova, Inorg. Mater., 2012, 48, 1279–1284 CrossRef CAS.
- B. K. Jena and C. R. Raj, Talanta, 2008, 76, 161–165 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |