
Physicochemical characterization of metal hexacyanometallate–TiO2 composite materials†
 *ab
*ab
Abstract
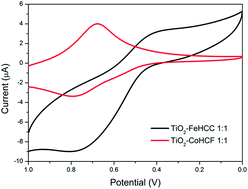
The paper describes the synthesis and characterization of novel TiO2–metal hexacyanometallates (MHCMs) composite materials. The starting material, TiO2, was modified by addition of cobalt-hexacyanoferrate (CoHCF) or iron-hexacyanocobaltate (FeHCC) at various concentrations. The resulting composites were characterized as follows: cyclic voltammetry (CV) followed the formation of TiO2–MHCM clusters, TEM micrographs studied their morphology, XAS and XPS data indicated that MHCM bonds to TiO2 through the nitrogen atom of its –CN group and modifies the environment of Ti compared to that of pure anatase. As expected, and confirmed by UV-Vis and XP-valence band data, the electronic properties of TiO2 were substantially modified: the edge in the composite materials shifted by about −2.0 eV relative to TiO2.
 Please wait while we load your content...
Please wait while we load your content...

