
Hyaluronic acid–PEI–cyclodextrin polyplexes: implications for in vitro and in vivo transfection efficiency and toxicity
S. Jain*a,
S. Kumarab,
A. K. Agrawala,
K. Thankia and
U. C. Banerjeeb
aCentre for Pharmaceutical Nanotechnology, Department of Pharmaceutics, National Institute of Pharmaceutical Education and Research (NIPER), Sector 67, S.A.S. Nagar, Mohali, Punjab 160062, India. E-mail: sanyogjain@niper.ac.in; sanyogjain@rediffmail.com; Fax: +91-172-2214692; Tel: +91-172-2292055
bDepartment of Pharmaceutical Technology, National Institute of Pharmaceutical Education and Research (NIPER), Sector 67, S.A.S. Nagar, Mohali, Punjab 160062, India
First published on 23rd April 2015
Abstract
The present study reveals novel HA–PEI–CyD polyplexes as non-viral vectors for gene delivery. The conjugate was synthesized and the phenol–sulfuric acid method revealed ∼5% and 14% grafting of cyclodextrin and hyaluronic acid, respectively, in the final conjugate. Model plasmid DNA, pEGFP-N3, was complexed with synthesized HA–PEI–CyD and a N/P ratio of 10 was found to be optimal and exhibited excellent stability in presence of serum and DNase I. In vitro transfection of HA–PEI–CyD polyplexes in HeLa, HEK-293 and MCF-7 cells revealed ∼39.5, 41.4 and 8.8 fold higher transfection as compared to plain PEI, respectively. Confocal laser scanning microscopy confirmed the higher cellular uptake and efficient nuclear colocalization of the HA–PEI–CyD polyplexes. An MTT assay revealed >90% cell viability in HA–PEI–CyD polyplexes in contrast to ∼20% cell viability in plain PEI. Furthermore, excellent GFP expression in tumor bearing animals and significantly lower in vivo toxicity along with hemocompatibility demonstrated the suitability of the proposed conjugate for gene delivery.
1. Introduction
Gene therapy has been evolved as a potential therapeutic strategy for various stubborn human diseases since last few decades. Therapeutic effectiveness and safety of gene therapy not only depends on the gene construct but also on the ability of the ferrying cargo to deliver the gene construct to its target site while avoiding deleterious consequences to normal cells. Immunogenicity and associated risk with the conventional viral vectors resulted in the emergence of better non-viral vectors as a potential alternative to them.1,2 Advantages like ease of preparation, being non-immunogenic, having the ability to shield the DNA against nucleases, a high DNA loading capacity, feasible attachment of the target ligand and low cost associated with non-viral vectors3–5 shifted the focus of the scientific community towards this germane area, which introduced a number of non-viral vectors viz. cationic polymers, cationic liposomes and dendrimers for gene delivery.1,2,6,7 These cationic vehicles electrostatically interact with negatively charged phosphate groups of DNA and condense DNA to submicron sized particles facilitating its cellular uptake.8Among the cationic polymers, polyethylenimine (PEI) has been recognized as a potential delivery reagent, primarily due to its excellent transfection efficiency assisted by the proton sponge effect and protection of DNA degradation against a harsh lysosomal environment.9 However, excessive cationic charge density on PEI results in interference with protein kinase C leading to apoptosis and cytotoxicity and a tendency to form larger aggregates with negatively charged blood components. This limits its use among the novel non-viral transfection reagents.10,11
Over time the PEGylation and coating of polysaccharides came into picture to modify the polycationic surface of PEI.12,13 Although PEGylated PEI exhibited reduced toxicity, no improvement was observed in transfection efficiency.14 In another approach, a polysaccharide coating was found to be superior as it imparted physicochemical stability along with site specificity, which resulted in improved transfection efficiency with reduced cytotoxicity.15,16 This concept was also observed and supported by our previous study, in which hyaluronic acid–PEI (HA–PEI) and chondroitin sulfate–PEI (CS–PEI) were found to have higher transfection efficiency and reduced cytotoxicity in comparison with parent PEI.17,18
Cyclodextrins (CyD) containing polycations has been explored as non-viral nanocarriers for gene therapy and exhibits higher transfection vis-à-vis lesser toxicity in comparison with non CyD containing polycations.19,20 Therefore, we hypothesized that the inclusion of CyD into HA–PEI and CS–PEI may further fortify the performance of these complexes. This presumption was based on the dual specification provided viz. the formation of an inclusion complex by guest host interaction, stability and target specificity due to CyD and HA, respectively. Thus, in the present study, HA–PEI–CyD based polyplexes were developed and evaluated for transfection efficiency, cellular uptake and nuclear colocalization and cytotoxicity in different cell lines. Finally, in vivo gene expression, toxicity in appropriate animal models and hemocompatibility were also checked.
2. Materials and methods
2.1 Materials
Branched PEI (MW 25 kDa), β-cyclodextrin (CyD), carbonyldiimidazole (CDI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), anhydrous DMF, Tris, bromophenol blue (BPB), ethidium bromide (EtBr), xylene cyanol (XC), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), rhodamine isothiocyanate (RITC), DMBA (7,12-dimethylbenz anthracene) were procured from Sigma-Aldrich Chemical Co., St. Louis, MO, USA. Dialysis membrane (MWCO 25 kDa) was procured from Spectrum Labs, USA. Hyaluronic acid (MW 5 kDa) was provided as generous gift sample from Focaschem Biotech limited, China. Plasmid (pEGFP-N3, 4.7 kb) encoded for enhanced green fluorescence protein was procured from Addgene. MCF-7 (human breast adenocarcinoma), HeLa (human cervix adenocarcinoma) and HEK-293 (human embryonic kidney-293) cell lines were obtained from the cell repository facility of the National Centre for Cell Sciences (NCCS), Pune, India. DNase was purchased from Fermentas Molecular Biology Tools. Deionized (MilliQ) and Millipore filtered (pore diameter 0.22 μm) water was used throughout the experiments. All other chemicals and reagents were of analytical grade and procured from local suppliers.2.2 Synthesis and characterization of HA–PEI–CyD
The synthesis of HA–PEI–CyD was carried out in two steps. In step 1, CyD–PEI backbone was synthesized by a previously reported method. Briefly, the hydroxyl groups of CyD were activated by dissolving CyD (0.015 mmol, 100 mg) in 20 mL anhydrous N,N-dimethylformamide (DMF) followed by dropwise addition of CDI (0.10 mmol). The mixture was stirred at room temperature for 1 h and then precipitated in cold diethylether and washed three times with diethylether to remove the unreacted materials. The resulting CyD–CDI was filtered and the residue was dissolved in 10 mL DMF and stored at 4 °C until use. PEI (0.010 mmol, 250 mg) was dissolved in DMF containing 100 μL of triethylamine (Et3N) followed by dropwise addition of CyD–CDI solution over 2 h with continuous stirring. The stirring was continued for additional 4 h to allow the reaction to complete. The mixture was then dialyzed overnight using dialysis tubing (MWCO, 25 kDa) in water and freeze-dried. In the next step, solutions of different concentration of HA (5, 10 and 15% w/v) in water were added to the aqueous solution of PEI–CyD followed by continuous stirring up to 24 h and dialysis for the next 24 h, which resulted in formation of HA–PEI–CyD1, HA–PEI–CyD2 and HA–PEI–CyD3, respectively.The concentration of CyD and HA in PEI–CyD and HA–PEI–CyD was determined using a phenol–sulphuric acid method.21 Briefly, 25 μL of PEI–CyD/HA–PEI–CyD (1 μg μL−1) was mixed thoroughly with 15 μL fresh 5% (w/v) phenol solution in double distilled water and 90 μL concentrated H2SO4 (95–97%) followed by incubation for 30 min at room temperature. Following incubation, samples were diluted up to 1 mL and the absorbance was recorded at 490 nm on a UV-VIS spectrophotometer (Shimadzu, UV-1800). Proton NMR spectroscopy was used to further characterize the synthesized complex. For analysis, 2 mg of complex was dissolved in 0.5 mL of D2O and a 1H NRM spectrum of each sample was recorded with a NMR spectrometer (400 MHz; Bruker Corporation). The synthesis of PEI–CyD and HA–PEI–CyD was confirmed by proton NMR spectroscopy.
2.3 Preparation and evaluation of HA–PEI–CyD/pDNA polyplexes
Polyplexes were formed at different N/P ratios (1.25, 2.5, 5, 7.5 and 10) by simply mixing the varying concentration of HA–PEI–CyD with pDNA solution in equivalent volumes followed by vortexing for 15 s and incubation at room temperature for 20 min. All the polyplexes had the final pDNA concentration of 2 μg mL−1. Herein, N/P ratio represents the ratio of the moles of the positively charged amino groups of cationic polymers to the moles of negatively charged phosphate groups of DNA. Polyplexes were characterized for size, polydispersity index (PDI) and zeta potential using zetasizer (Nano ZS, Malvern, UK). The shape and morphology of the optimized complex was studied using scanning electron microscopy (SEM, Hitachi, S-3400N, Japan) after gold coating and atomic force microscopy (AFM, Veeco, di Bioscope SZ).22–242.4 Optimization of N/P ratio: agarose gel electrophoresis
For gel electrophoresis, polyplexes were gently mixed with loading buffer containing xylene cyanol, a tracking dye, and loaded into individual wells of 0.8% agarose gel and electrophoresed at 100 V for 45 min in TAE buffer (40 mM Tris–HCl, 1% (v/v) acetic acid, 1 mM EDTA). The gels were stained using ethidium bromide (EtBr) and the bands corresponding to pDNA were visualized under a UV transilluminator.17,182.5 DNase protection assay
The ability of the developed polyplexes to protect the complexed DNA against DNase-I was evaluated by the DNase protection assay.25 Briefly, naked plasmid DNA and different polyplexs (formed at N/P ratio 10) were incubated at 37 °C for 30 min in a buffer solution (10 × 10−3 M Tris–Cl, 150 × 10−3 M NaCl and 1 × 10−3 M MgCl2, pH 7.4) containing 10 μL DNase I (1000 units mL−1). In addition, 50 μL of Mg+2 solution (50 × 10−3 M) was also added to initiate the enzymatic reaction. The change in absorbance value was measured spectrophotometrically at 260 nm at an interval of 2 min.In another method, protection against DNase I was confirmed by gel electrophoresis. The procedure was same as described above followed by the inactivation of DNase I with the addition of 5 μL 100 mM EDTA (10 min). Subsequently, the complexes were incubated in the presence of 10 μL heparin (5 mg mL−1) for 2 h to dissociate the complexes. The samples were electrophoresed in 0.8% agarose to examine pDNA replaced from the complex.
2.6 Serum stability
In vitro serum stability of the developed polyplexes was determined by measuring changes in the critical quality attributes (particle size and zeta potential) and by an EtBr intercalation assay.26 Briefly, naked plasmid DNA and different polyplexes (at N/P ratio 10) were incubated at 37 °C for different time points (0.5, 1, 2 and 4 h) with an equal volume of PBS buffer (pH 7.4) containing 20% FBS to have 10% of FBS in the final mixture. Following incubation, samples (1 mL) at corresponding time points were mixed with EtBr solution in water to get the final concentration, 0.2 μg mL−1 of EtBr. The fluorescence intensity of the resultant mixtures was determined with 516 nm excitation wavelength and a 618 nm emission wavelength. The % change in EtBr fluorescence was calculated as a fraction of observed fluorescence to the maximum fluorescence, obtained upon the addition of EtBr to the free plasmid. The sample collected after 4 h of incubation was also examined for change in the particle size and zeta potential before processing for the EtBr intercalation assay.2.7 In vitro transfection, cytotoxicity, cellular uptake and colocalization studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. 2 μL of the supernatant was loaded on a Nanodrop spectrofluorimeter (NanoDrop 3300, Thermo Fisher Scientific, USA) and EGFP expression was estimated fluorimetrically at excitation and emission wavelengths of 489 and 510 nm, respectively. EGFP expression of the polyplexes was also measured against blank cells (without pDNA).17,18
000 rpm for 10 min. 2 μL of the supernatant was loaded on a Nanodrop spectrofluorimeter (NanoDrop 3300, Thermo Fisher Scientific, USA) and EGFP expression was estimated fluorimetrically at excitation and emission wavelengths of 489 and 510 nm, respectively. EGFP expression of the polyplexes was also measured against blank cells (without pDNA).17,18| % Relative cell viability = 100 × Abs(sample)/Abs(control) |
Following overnight incubation, different cells, MCF-7, HeLa and HEK-293, were allowed to incubate with RITC incorporated polyplexes for 4 h. Following 4 h of incubation, the cells were thoroughly washed with PBS to remove the non-internalized complexes and were fixed by adding 4% paraformaldehyde in PBS (pH 7.4) for 10 minutes. After fixing, the cells were thoroughly washed with PBS and subjected to Triton X-100 (0.2% in PBS) for 2 min for permeabilization. The cell nuclei were counterstained with DAPI (1 μg mL−1 in PBS) for 1 min followed by thorough washing with PBS.27 The cells were visualized in CLSM (Olympus FV1000, Japan) at excitation/emission maxima of ∼358/461 nm for DAPI and ∼560/580 nm maxima for RITC. Scatter plot analysis was performed for nuclear colocalization studies.
2.8 In vitro hemocompatibility
Blood was collected from rats in heparinized microcentrifuge tubes and subjected to centrifugation at 3000 rcf for 5 min at 4 °C to separate the red blood cells (RBCs). The supernatant was discarded and the RBCs were washed thrice with isotonic PBS at pH 7.4. The stock of RBCs was prepared by mixing three volumes of RBCs with 11 parts of PBS. 100 μL of this stock was mixed with PEI, PEI–CyD and PEI–CyD–HA complexes, which is equivalent to 10 μg mL−1 of PEI. RBCs, mixed with distilled water and PBS, were employed as positive and negative controls, respectively. The samples were incubated at 37 °C for 3 h in a shaker bath and then centrifuged at 3000 rcf for 5 min to separate the supernatant, which was allowed to stand at room temperature for 30 min to oxidize hemoglobin (Hb). The absorbance of oxygenated hemoglobin (Oxy-Hb) was measured spectrophotometrically at 540 nm, and the percentage hemolysis was calculated using the following equation:where ABs is the absorbance of the sample and AB100 is the absorbance of the control.

2.9 In vivo studies
2.10 Statistical analysis
All results have been presented as mean ± standard deviation (SD). Statistical analysis was performed using Graph Pad Prism 6 using the one-way analysis of variance (ANOVA) followed by a Tukey–Kramer multiple comparison test. p < 0.05 was considered to be statistically significant.3. Results
3.1 Synthesis and characterization of HA–PEI–CyD
The synthesis of PEI–CyD and HA–PEI–CyD was confirmed by proton NMR spectroscopy (Fig. 1).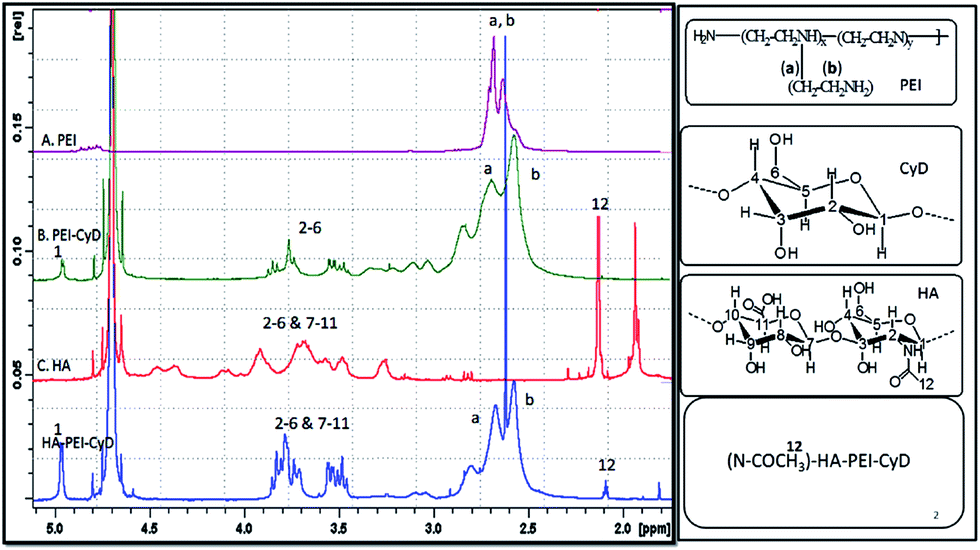 | ||
| Fig. 1 1HNMR of PEI, PEI–CyD, HA and HA–PEI–CyD. | ||
The signals from PEI ethylene protons (–CH2CH2NH–) appeared at δ 2.5–3.0 ppm, while the C1 proton and C2–C6 protons of β-CyD appeared at δ 5 ppm and δ 3.0–4.0 ppm, respectively. The formation of HA–PEI–CyD was confirmed by the presence of apparent N-acetylate protons (–NCOCH3–) of HA at δ 2.1 ppm. The other proton peaks that ranged between δ 3.5 and 4.0 ppm could not be distinguished because of the overlap of the glucose unit peak of HA and CyD. The % w/w grafting of CyD in PEI–CyD and HA in the corresponding HA–PEI–CyD are shown in Table 1.
| CyD grafting | HA grafting | |
|---|---|---|
| PEI | — | — |
| PEI–CyD | 4.8 | — |
| HA–PEI–CyD1 | 4.8 | 4.3 |
| HA–PEI–CyD2 | 4.8 | 9.4 |
| HA–PEI–CyD3 | 4.8 | 14.2 |
The phenol–sulfuric acid method revealed 5% and ∼14% w/w grafting of CyD and HA in the final complexes.
3.2 Preparation and evaluation of HA–PEI–CyD/pDNA polyplexes
The particle size and zeta potential at different N/P ratios for the polyplexes are shown in Table 2.| N/P | PEI | PEI–CyD | HA–PEI–CyD1 | HA–PEI–CyD2 | HA–PEI–CyD3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Size (nm) | Zeta potential (mV) | Size (nm) | Zeta potential (mV) | Size (nm) | Zeta potential (mV) | Size (nm) | Zeta potential (mV) | Size (nm) | Zeta potential (mV) | |
| a All values are expressed as mean ± SD (n = 6). | ||||||||||
| 1.25 | 437 ± 23 | 12.1 ± 1.7 | 472 ± 19 | 6.2 ± 1.1 | 412 ± 19 | 3.4 ± 1.9 | 398 ± 17 | 2.27 ± 1.8 | 367 ± 12 | 1.3 ± 1.1 |
| 2.5 | 239 ± 17 | 22.9 ± 1.3 | 376 ± 16 | 18.3 ± 1.7 | 285 ± 16 | 9.2 ± 1.4 | 261 ± 11 | 7.93 ± 1.2 | 239 ± 9 | 3.4 ± 1.5 |
| 5 | 201 ± 7 | 36.3 ± 1.3 | 281 ± 12 | 33.1 ± 1.7 | 271 ± 12 | 24.1 ± 1.1 | 252 ± 9 | 16.7 ± 1.3 | 237 ± 8 | 13.9 ± 1.4 |
| 7.5 | 193 ± 13 | 35.1 ± 1.1 | 268 ± 14 | 29.3 ± 1.2 | 259 ± 15 | 19.6 ± 1.6 | 237 ± 8 | 15.1 ± 1.7 | 219 ± 11 | 12.6 ± 1.2 |
| 10 | 189 ± 16 | 33.7 ± 1.4 | 254 ± 13 | 26.1 ± 1.9 | 247 ± 18 | 16.1 ± 1.2 | 223 ± 11 | 13.6 ± 1.5 | 201 ± 9 | 10.8 ± 1.4 |
In all the polyplexes, particle size was reduced upon increasing the N/P ratio from 1.25 to 10. The difference in various formulation parameters was not significant between the N/P ratios 7.5 and 10. Zeta potential was invariably increased upon increasing the N/P ratio; however, the zeta potential of PEI–CyD was lower compared to PEI polyplexes alone. The zeta potential was further reduced in the case of HA–PEI–CyD polyplexes upon increasing the HA content from 5% to 15%. The polyplexes were found to have an almost spherical shape (Fig. 2A and B).
 | ||
| Fig. 2 Shape and morphology of HA–PEI–CyD1 at N/P ratio 10 (A) SEM (B) AFM. | ||
3.3 Agarose gel electrophoresis
A complete retardation of electrophoretic mobility was observed at a N/P ratio 1.25 in case of PEI polyplexes, while this was shifted towards higher N/P ratios in the case of CyD and HA conjugated polyplexes (Fig. 3). | ||
| Fig. 3 Agarose gel electrophoresis. Lane 1 represents uncomplexed pDNA, while Lane 2–6 represents polyplexes at 1.25, 2.5, 5, 7.5 and 10, respectively. | ||
However, complete electrophoretic retardation was observed at a N/P ratio of 7.5 for all the complexes yet an N/P ratio of 10 was implemented for further studies based on our previous experience (Table 2).
3.4 DNase protection assay
Cleavage of DNA in the presence of DNase results in the formation of nucleotide, which was confirmed by an increased absorbance at 260 nm. The change in absorbance for different polyplexes is shown in Fig. 4. | ||
| Fig. 4 DNase protection assay. Plot indicates change in absorbance with time. | ||
Naked pDNA cleaved to nucleotides in the presence of DNase, as evident by the increased absorbance at 260 nm that was observed up to 10 min. Different polyplexes were able to protect the complexed pDNA, as no change in the absorbance value was observed at different time points. Although the protection efficiency of all polyplexes was significantly higher (p < 0.001) in comparison with naked pDNA, the difference was insignificant (p > 0.05) among the different polyplexes. The results clearly indicated that pDNA complexed with PEI was resistant to nuclease attack irrespective of the presence of CyD and HA in the PEI backbone.
The electrophoretic movement of pDNA is shown in Fig. 2B. No electrophoretic mobility was observed in case of naked pDNA incubated in the presence of DNase, while the electrophoretic mobility was retained (Fig. 5, lane 3–7) in the case of different polyplexes.
 | ||
| Fig. 5 DNase protection assay. Lane 1: pDNA without DNase treatment; lane 2: pDNA incubated with DNase; lane 3 and 4: PEI and PEI–CyD; lane 5–7: HA–PEI–CyD (1–3). | ||
3.5 Serum stability
| Formulation | Size (nm) | Zeta potential (mV) | |||
|---|---|---|---|---|---|
| Before | After | Before | After | ||
| a Values are expressed as mean ± SD (n = 6). | |||||
| PEI | 189 ± 16 | 557 ± 18 | +33.7 ± 1.4 | −24.3 ± 1.6 | |
| PEI–CyD | 254 ± 13 | 457 ± 31 | +26.1 ± 1.9 | +4.4 ± 1.2 | |
| HA–PEI–CyD1 | 247 ± 18 | 268 ± 19 | +16.1 ± 1.2 | +12.1 ± 1.3 | |
| HA–PEI–CyD2 | 223 ± 11 | 247 ± 11 | +13.6 ± 1.5 | +10.5 ± 1.8 | |
| HA–PEI–CyD3 | 201 ± 9 | 223 ± 9 | +10.8 ± 1.4 | +7.6 ± 1.4 | |
A drastic increase in size and reduction of the zeta potential was observed in the case of PEI and PEI–CyD polyplexes, while all the HA–PEI–CyD (1–3) polyplexes were found to be quite stable, as only a slight increase in size and decrease in zeta potential were observed.
 | ||
| Fig. 6 Stability of different polyplexes in the presence of serum. Plot indicates the % change in EtBr fluorescence with respect to time. | ||
3.6 In vitro transfection efficiency
The in vitro transfection efficiency of the developed non viral vectors was assessed as a function of qualitative and quantitative tools. Confocal images of the cells treated with various transfecting agents and developed novel vectors revealed that HA modified complexes exhibited remarkably higher fluorescence in comparison with PEI and PEI–CyD in all the cell lines. However, the fluorescence observed in HeLa and HEK-293 cell lines was much higher in comparison with MCF-7 cell lines (Fig. 7).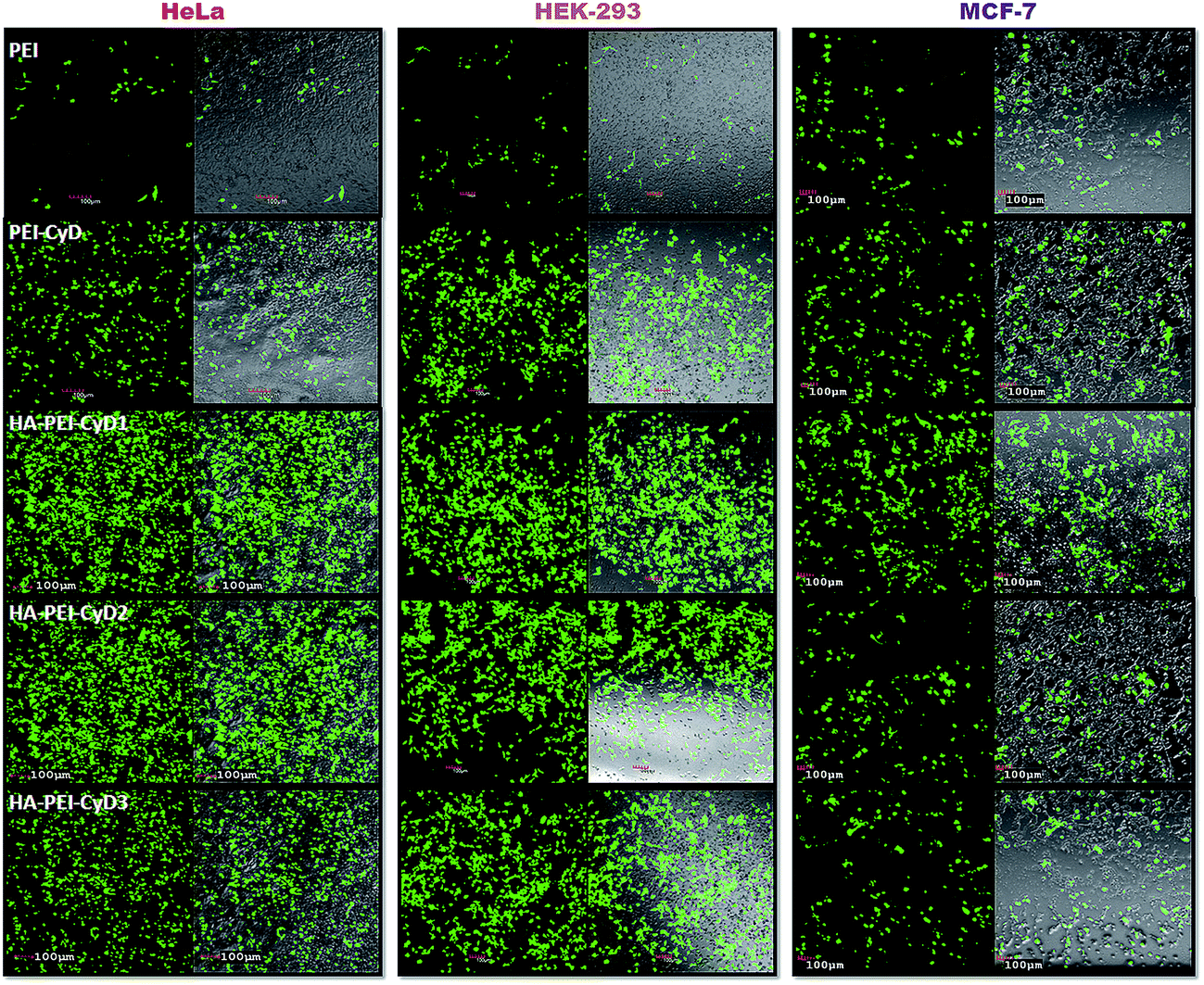 | ||
| Fig. 7 CLSM images of HeLa, HEK-293 and MCF-7 cells. For each incubation type, the left and right panels represent GFP fluorescence (green) and the overly of GFP and DIC (Differential interference contrast image). | ||
Concomitantly, similar results were also noted in case of flow cytometry assessment wherein >80% cell transfection was noted for HA–PEI–CyD in HeLa and HEK 293 cells. In contrast, ∼20% and ∼40% transfection efficiency was noted in the cases of PEI and PEI–CyD, respectively, in all the cell lines. Interestingly, in case of HA–PEI–CyD, the lower transfection efficiency (∼50%) was noted in the MCF-7 cell line, which was almost comparable with that of PEI–CyD (∼40%) (Fig. 8). The results could be attributed to the relative overexpression of CD44 receptors on the HeLa and HEK 293 cell lines as compared to that of the MCF-7 cells. Furthermore, the quantitative estimation of cell transfection using a fluorescence spectrophotometer suggested 4.1, 6.5 and 5.7 fold enhanced transfection in comparison with PEI alone in HeLa, HEK-293 and MCF-7, respectively, upon the inclusion of CyD to the PEI backbone. The highest transfection was observed in the case of HA–PEI–CyD1, which was 39.5, 41.5 and 8.8 fold higher as compared to PEI, while 9.6, 6.3 and 1.4 fold higher as compared to the PEI–CyD in the HeLa, HEK-293 and MCF-7 cell lines, respectively (Fig. 9).
 | ||
| Fig. 8 Flow cytometry based assessment of in vitro transfection efficiency in (A) HeLA cell lines, (B) HEK 293 cell lines and (C) MCF-7 cell lines using the PEI, PEI–CyD and HA–PEI–CyD polyplexes. | ||
 | ||
| Fig. 9 Transfection efficiency of PEI, PEI–CyD and HA–PEI–CyD polyplexes in different cell lines. | ||
3.7 Cytotoxicity assay
Percent cell viability in case of PEI polyplexes was found to be <25%, while it was ≥70% in the case of PEI–CyD in all the cell lines. Further modification with HA resulted in >90% cell viability in the case of HA–PEI–CyD polyplexes, which was significantly higher in comparison with PEI (p < 0.001) and PEI–CyD (P < 0.05). Although the percentage of cell viability slightly increased upon increasing the HA concentration, it was still insignificant (p > 0.05) (Fig. 10).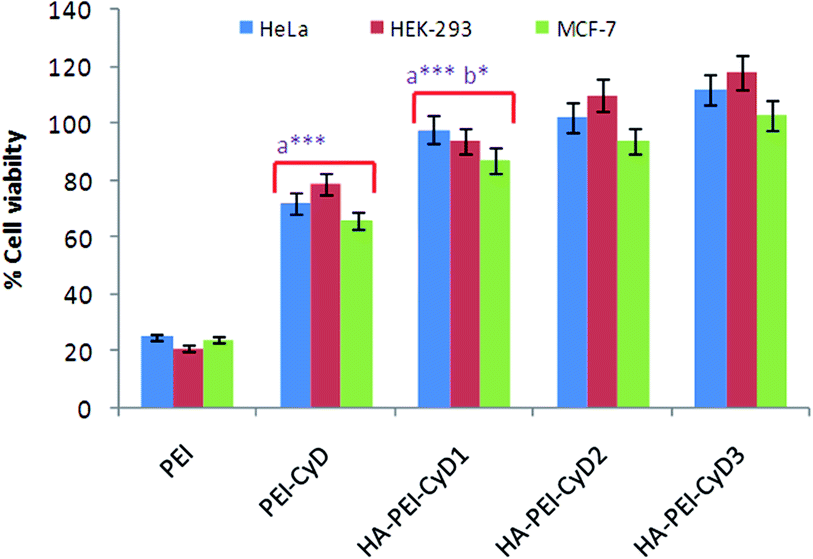 | ||
| Fig. 10 In vitro cell viability of HeLa, HEK-293 and MCF-7 cells incubated with various polyplexes ((a) in comparison with PEI, (b) in comparison with PEI–CyD) (*** p < 0.001, * p < 0.05). | ||
3.8 Cell uptake and colocalization studies
For cell uptake and colocalization, HA–PEI–CyD1 was taken into consideration as it exhibited the highest transfection and was compared with PEI and PEI–CyD as basic and intermediate products. Representative confocal images of HeLa, HEK-293 and MCF-7, cells incubated with different complexes are shown in Fig. 11. | ||
| Fig. 11 Confocal microscopic images of HeLa, HEK-293 and MCF-7 cell lines treated with RITC-complexes. The white line represents the scale bar of 10 nm. The left, middle and right panel of each incubation type represents RITC fluorescence, overlay of RITC and DAPI fluorescence and scatter plot analysis for the entire field of view. The horizontal and vertical axes of each scatter plot represent the values of pixels in channel 2 (ch2) and channel 1 (ch1), respectively. | ||
As evident from the images, HA modified complex exhibited qualitatively a very high internalization in all the three cell lines. Moreover, the highest colocalization coefficient was observed in case of HA modified complexes (r > 0.5 in all the cell lines), followed by PEI–CyD and PEI polyplexes.
3.9 In vitro hemocompatibility
Hematological parameters were affected significantly in case of PEI, while it was insignificant in case of developed polyplexes (data not shown). Hemolytic toxicity profile and % hemolysis observed in case of different complexes is shown in Fig. 12A and B, respectively. | ||
| Fig. 12 Hemocompatibility of different complexes (A) color produced as a result of hemoglobin oxidation (B) percent hemolysis. (*** p < 0.001, ** p < 0.01) (n = 3). | ||
Complete lysis was observed in case of the positive control (distilled water) and considered 100%, while no hemolysis was observed in case of the negative control (PBS, pH 7.4). Incubation of RBCs with PEI led to significant hemolysis (P < 0.001) as compared to PEI–CyD and HA–PEI–CyD. Furthermore, PEI–CyD also showed significant (p < 0.01) hemolysis as compared to HA–PEI–CyD.
3.10 In vivo gene expression
In order to confirm the delivery potential of the designed conjugate, an in vivo gene expression study was designed in which different conjugates as well as free DNA were injected intratumorally and green fluorescence was produced as a result of gene expression was observed. Intense fluorescence was observed in all the complexes, while fluorescence was negligible in the case of free DNA (Fig. 13).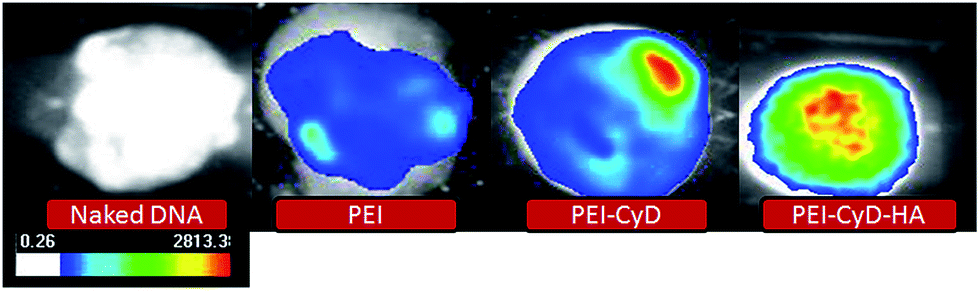 | ||
| Fig. 13 Images of the excised tumors showing GFP expression following intratumoral injection of naked plasmid DNA and different polyplexes in tumor bearing rats. | ||
3.11 In vivo toxicity
Changes in different biochemical markers following the administration of different polyplexes are shown in Fig. 14.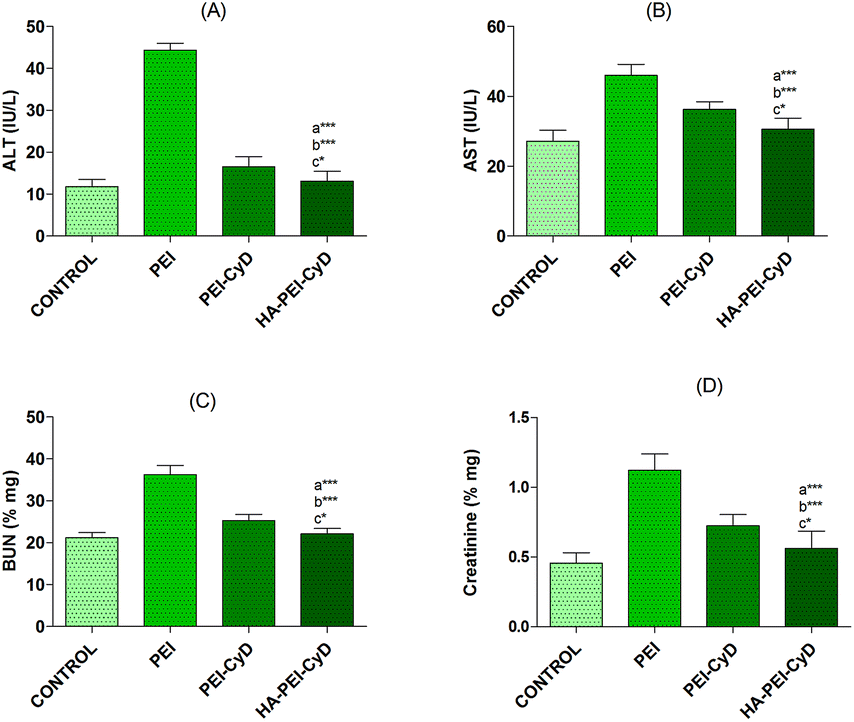 | ||
| Fig. 14 Levels of serum biochemical markers (A) ALT (B) AST (C) BUN (D) creatinine following intravenous administration of PEI, PEI–CyD and HA–PEI–CyD polyplexes. | ||
No significant difference in the levels of various biochemical parameters viz. ALT and AST for liver function (Fig. 14A and B), and BUN and creatinine (Fig. 14C and D) for renal function was observed in the case of HA–PEI–CyD, while they were significantly changed (P < 0.001) in the case of the PEI and PEI–CyD polyplexes compared with the control.
4. Discussion
In the present study, HA–PEI–CyD based polyplexes were developed with the assumption that the inclusion of CyD and HA would provide dual specification and fortify the efficacy of the developed system. For this purpose, HA–PEI–CyD was synthesized and characterized by proton NMR spectroscopy. Following synthesis, the polyplexes were formed at different N/P ratios by incubating the synthesized conjugate with DNA. The formation of polyplexes was based on the principle of electrostatic attraction between the positively charged amino groups of HA–PEI–CyD and the negatively charged phosphate groups of DNA. The size and surface charge of the particles play an important role in cellular uptake as the positively charged complex interacts with the negatively charged proteoglycans of the cell membrane, which results in enhanced cellular uptake over neutral and negatively charged complexes. In all the polyplexes, the particle size was reduced, which could be attributed to the condensation of the negatively charged plasmid DNA with increasing the amount of the cationic counterpart. Among the different N/P ratios tested, the minimum particle size observed at N/P ratios 7.5 and 10 may have been the consequence of formation of the most condensed structure due to the balance between the positive and negative charge moieties. Higher size at N/P ratios >10 could be ascribed to the formation of loosely packed aggregates due to excessive positive charge (data not shown). Conjugation of CyD to PEI resulted in a larger complex, while increasing the concentration of HA resulted in the formation of small sized complexes. The results are in agreement with our previous observation and could be attributed to the formation of more condensed and spherical structures due to strong electrostatic attraction.17 Invariable increase in zeta potential upon increasing the N/P ratio could be ascribed to the obvious reason of a higher concentration of the cationic counterpart. The comparatively lower zeta potential of PEI–CyD and HA–PEI–CyD to PEI polyplexes might be the consequence of some degree of the capping of positively charged amino groups by CyD. The results are in agreement with previous reports.29Conjugation of PEI with CyD and HA can negatively influence the complexation efficiency of PEI to pDNA. In order to determine the effect of conjugation of PEI with CyD and HA, agarose gel electrophoresis was performed at different N/P ratios (1.25, 2.5, 5, 7.5 and 10). Shifting of the N/P ratio could be attributed to the some degree of capping of a fraction of positively charged amino groups of PEI by bulky CyD and HA, rendering them inaccessible for binding to DNA. This was further confirmed by shifting the N/P ratio towards the higher side upon increasing the HA concentration from HA–PEI–CyD (1–3). A higher concentration of HA might have blocked more amino groups and therefore reduced their availability for complexation with pDNA.
The DNase assay is based on the principle of degradation of free DNA in the presence of DNase. An ideal gene delivery reagent is expected to provide protection against nuclease present in different biological fluids. Stability of complexed pDNA in polyplexes could be attributed to the strong electrostatic attraction between positively charged amine groups of PEI and negatively charged phosphate groups of pDNA.
In another approach, the protection of pDNA complexed in different polyplexes was confirmed by gel electrophoresis. Incubation of polyplexes with heparin solution resulted in the displacement of pDNA from the complex due to the high binding efficiency of heparin to PEI compared with pDNA. This displaced free pDNA exhibited electrophoretic mobility in gel electrophoresis. No electrophoretic mobility in case of naked pDNA could be attributed to the degradation of pDNA and complete loss of electrophoretic mobility, while the retention of electrophoretic mobility in case of different complexes might be the consequence of protection of complexed pDNA against DNase I. The results of gel electrophoresis further supported our previous observation in which the protection efficiency of complexes was evaluated by measuring the change in absorbance.
Serum stability of polyplexes is an important parameter, which directly influences the in vivo performance of the polyplexes. Cationic PEI polyplexes are known to interact with anionic proteins in the serum, which results in the formation of larger sized aggregates. This ultimately leads to reduced endocytosis and poor transfection efficiency.30 Reduction of protein adsorption by several means has been shown to improve transfection efficiency.31 Therefore, serum stability was taken as a quality control tool to determine the stability of developed complexes in the presence of serum. A drastic increase in size and reduction in zeta potential observed in case of PEI and PEI–CyD polyplexes could be attributed to the adsorption of negatively charged serum proteins over the positively charged polyplexes due to strong electrostatic attraction. This observation was in agreement with previous reports.32 Improved stability of all the HA–PEI–CyD (1–3) polyplexes could be contributed by the partial neutralization of excessive positive charge by HA, which ultimately decreased the tendency of protein adsorption due to reduced electrostatic attraction. Along with changes in their physicochemical characteristics, negatively charged proteins can also displace the complexed DNA. For this purpose, the EtBr intercalation assay was performed, which is based on the principle of intercalation of EtBr between the base pairs of double helix of displaced DNA, resulting in emission of an intense fluorescence signal at 618 nm when excited at 516 nm. High charge density over the PEI and PEI–CyD could provide strong electrostatic attraction with serum proteins, which might have led to the displacement of pDNA from the complex and destabilization of these complexes. Surface modification of PEI using different concentrations of HA resulted in the masking of excessive positive charge, which could stabilize the system in the presence of serum by reducing excessive adsorption of serum proteins.
The transfection efficiency of a gene delivery reagent not only depends upon the purity of the gene construct but is also equally contributed by the efficacy and target specificity of the delivery vehicle. Furthermore, specialized uptake mechanisms and over expression of specific receptors on different cells necessitates the design of surface engineered delivery reagents, which can deliver the gene construct more specifically to target cells. To provide target specificity, HA was appended on PEI–CyD backbone, which binds specifically to CD44 receptors. These receptors have been reported to over express on a variety of tumor cells viz. breast, colon, bowel, brain, melanoma, sarcoma, renal and prostate.17 To exactly mimic and evaluate the effect of over expression, HeLa, HEK-293 and MCF-7 cell lines were used to estimate in vitro transfection efficiency. Selection of cell lines was based on the level of expression of CD44 receptors, as CD44 shows over expression in HeLa and HEK-293,17,18 while low level of expression in MCF-7.33 Higher fluorescence observed in the case of HA modified PEI–CyD could be attributed to the enhanced uptake of the polyplexes through receptor mediated endocytosis. Furthermore, quite higher fluorescence in the case of HeLa and HEK-293 in comparison with MCF-7 might be the consequence of over-expression of HA receptors in the HeLa and HEK-293 cell lines and a low level of expression of MCF-7. In line with previous reports, the inclusion of CyD to the PEI backbone could fortify the transfection efficiency.29 CyDs are cup-shaped cyclic oligomers of glucose, which have been well recognized for their importance in gene transfection. By forming inclusion complexes via a host–guest interaction, CyDs provide facile and versatile attachment sites for ligand anchorage.29 Based on the outcomes of our previous study, we speculated that the inclusion of CyD can improve the efficacy of targeted transfection by providing multiple attachment sites to HA. As both HA and pDNA are negatively charged, they can compete with each other for binding to PEI and can adversely affect the performance. Therefore, the attachment of HA as an inclusion complex can prevent such unnecessary competition. HA modification could result into improved transfection, while diminished transfection observed at higher concentrations of HA (HA–PEI–CyD2 and HA–PEI–CyD3) could be attributed to the masking of additional charge of PEI by excess HA. At a low concentration (5%), almost entire HA could be utilized to form inclusion complexes without affecting the positively charged amino groups of PEI much. In support of our hypothesis, HA–PEI–CyD exhibited significantly higher transfection efficiency in comparison with our previously reported HA–PEI polyplexes.17
Although PEI based complexes have been recognized for their tremendous potential in gene delivery, they are fraught with the major limitation of higher cell cytotoxicity due to the excessive positive charge density of these systems. Excessive positive charge and molecular weight are two major factors, which have been recognized to exhibit high cytotoxicity.17,18 In order to nullify the excessive charge, PEI was first modified with CyD to obtain PEI–CyD that was further modified by anchoring the targeting ligand HA to PEI–CyD backbone. The significantly higher cell viability observed in the case of HA–PEI–CyD could be attributed to the capping of the additional charge of PEI by CyD and HA. Lower toxicity at a higher concentration of HA could be ascribed to the same reason of neutralization of charge on PEI.
In order to understand the underlying mechanism behind the much higher transfection in HA modified complexes, comparative cell uptake and colocalization studies were performed using CLSM and flow cytometry. Significantly higher internalization could be attributed to the receptor mediated uptake due to HA modification, which facilitated their binding and internalization through interaction with CD44 receptors on cell membranes.17 By this observation, we speculated that this higher transfection may be the result of efficient nuclear colocalization of the HA modified complex. Thus, our next step was to elucidate the delivery efficiency of the HA modified complex to deliver the loaded DNA into the site of transfection, i.e. nucleus. A colocalization coefficient greater than or equal to 0.5 (r ≥ 0.5) is usually considered as an indicator of efficient colocalization.34 A good colocalization coefficient observed in our case can be rationalized by concentrating on the stability profile of an individual formulation in the presence of serum. As evident from Table 3, PEI-polyplexes displayed negative charges in the presence of serum, while it was reduced noticeably in case of PEI–CyD polyplexes. HA modified complexes were quite stable as no significant change in size and charge was observed. Thus, amongst all the investigated complexes, only HA modified complexes are supposed to retain sufficient positive charges in the intracellular milieu to get internalized and rapid endolysosomal escape via the proton sponge effect and subsequent colocalization in the nucleus.
In line with our concept, the synthesized conjugate demonstrated better compatibility in comparison to PEI and PEI–CyD polyplexes and this could be directly related to capping of excess cationic groups of PEI. Furthermore, in order to confirm the delivery potential of the designed conjugate, an in vivo gene expression study was designed in which different conjugates as well as free DNA were injected intratumorally and green fluorescence was produced as a result of gene expression was observed. In line with our hypothesis, the HA–PEI–CyD expressed much higher fluorescence in comparison with PEI and PEI–CyD complexes. The enhanced efficacy observed in case of HA–PEI–CyD could be correlated with our in vitro results in which HA modification resulted in improved stability in comparison to other complexes. Furthermore, receptor mediated uptake due to HA modification and rapid endolysosomal escape via the proton sponge effect and subsequent colocalization in the nucleus could improve the overall performance.
In order to further prove the improved safety profile of the developed polyplexes, in vivo toxicity studies were performed. These in vivo findings are quite consistent with our previous observations of cytotoxicity assay on different cell lines and further strengthen the concept that the masking of excessive charge of PEI can be utilized as a fruitful strategy to overcome toxicity related issues. Simultaneously, careful selection of the compounds in system fabrication can be used to control the intracellular trafficking as desirable.
5. Conclusions
The proposed HA–PEI–CyD was found to have better transfection efficiency and lower cytotoxicity in comparison with PEI–CyD and previously reported HA–PEI in different cell lines, which strongly supports the suitability of the system in designing reagent delivery with target specificity. The system further provides an insight to develop a variety of gene delivery constructs by considering different targeting ligands. Encouraged with these findings, we are at the stage of evaluating in vivo bio-distribution and gene expression following intravenous administration, which will be reported over time.Acknowledgements
Authors are thankful to Indian National Science Academy (INSA), Government of India, New Delhi, for providing financial assistance, the Council of Scientific and Industrial Research (CSIR) Govt. of India, India for providing fellowship to Mr AKA and KT and Director, NIPER for providing the necessary infrastructure facilities.References
- S. C. De Smedt, J. Demeester and W. E. Hennink, Pharm. Res., 2000, 17, 113–126 CrossRef CAS.
- S. A. Audouy, L. F. de Leij, D. Hoekstra and G. Molema, Pharm. Res., 2002, 19, 1599–1605 CrossRef CAS.
- D. J. Glover, H. J. Lipps and D. A. Jans, Nat. Rev. Genet., 2005, 6, 299–310 CrossRef CAS PubMed.
- G. D. Schmidt-Wolf and I. G. Schmidt-Wolf, Trends Mol. Med., 2003, 9, 67–72 CrossRef CAS.
- T. Bronich, Pharm. Res., 2010, 27, 2257–2259 CrossRef CAS PubMed.
- B. H. Zinselmeyer, S. P. Mackay, A. G. Schatzlein and I. F. Uchegbu, Pharm. Res., 2002, 19, 960–967 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- V. Vijayanathan, T. Thomas and T. J. Thomas, Biochemistry, 2002, 41, 14085–14094 CrossRef CAS PubMed.
- H. Aldawsari, B. S. Raj, R. Edrada-Ebel, D. R. Blatchford, R. J. Tate, L. Tetley and C. Dufes, Nanomedicine, 2011, 7, 615–623 CrossRef CAS PubMed.
- D. Jiang and A. K. Salem, Int. J. Pharm., 2012, 427, 71–79 CrossRef CAS PubMed.
- C. H. Ahn, S. Y. Chae, Y. H. Bae and S. W. Kim, J. Controlled Release, 2002, 80, 273–282 CrossRef CAS.
- B. Liang, M. L. He, C. Chan, Y. Chen, X. P. Li, Y. Li, D. Zheng, M. C. Lin, H. F. Kung and X. T. Shuai, Biomaterials, 2009, 30, 4014–4020 CrossRef CAS PubMed.
- S. Kommareddy and M. Amiji, Nanomedicine, 2007, 3, 32–42 CrossRef CAS PubMed.
- G. P. Tang, J. M. Zeng, S. J. Gao, Y. X. Ma, L. Shi, Y. Li, H. P. Too and S. Wang, Biomaterials, 2003, 24, 2351–2362 CrossRef CAS.
- S. Kawakami, A. Sato, M. Nishikawa, F. Yamashita and M. Hashida, Gene Ther., 2000, 7, 292–299 CrossRef CAS PubMed.
- S. Patnaik, A. Aggarwal, S. Nimesh, A. Goel, M. Ganguli, N. Saini, Y. Singh and K. Gupta, J. Controlled Release, 2006, 114, 398–409 CrossRef CAS PubMed.
- A. Pathak, A. Swami, S. Patnaik, S. Jain, K. Chuttani, A. K. Mishra, S. P. Vyas, P. Kumar and K. C. Gupta, J. Biomed. Nanotechnol., 2009, 5, 264–277 CrossRef CAS PubMed.
- A. Pathak, P. Kumar, K. Chuttani, S. Jain, A. K. Mishra, S. P. Vyas and K. C. Gupta, ACS Nano, 2009, 3, 1493–1505 CrossRef CAS PubMed.
- H. Yao, S. S. Ng, W. O. Tucker, Y. K. T. Tsang, K. Man, X. Wang, B. K. C. Chow, H. F. Kung, G. P. Tang and M. C. Lin, Biomaterials, 2009, 30, 5793–5803 CrossRef CAS PubMed.
- S. H. Pun, N. C. Bellocq, A. Liu, G. Jensen, T. Machemer, E. Quijano, T. Schluep, S. Wen, H. Engler, J. Heidel and M. E. Davis, Bioconjugate Chem., 2004, 15, 831–840 CrossRef CAS PubMed.
- X. Q. Zhang, X. L. Wang, P. C. Zhang, Z. L. Liu, R. X. Zhuo, H. Q. Mao and K. W. Leong, J. Controlled Release, 2005, 102, 749–763 CrossRef CAS PubMed.
- S. Jain, S. Kumar, A. K. Agrawal, K. Thanki and U. C. Banerjee, Mol. Pharmaceutics, 2013, 10, 2416–2425 CrossRef CAS PubMed.
- S. Jain, S. R. Patil, N. K. Swarnakar and A. K. Agrawal, Mol. Pharmaceutics, 2012, 9, 2626–2635 CrossRef CAS PubMed.
- A. K. Agrawal, H. Harde, K. Thanki and S. Jain, Biomacromolecules, 2014, 15, 350–360 CrossRef CAS PubMed.
- S. H. Kim, J. H. Jeong, C. O. Joe and T. G. Park, J. Controlled Release, 2005, 103, 625–634 CrossRef CAS PubMed.
- S. Li, W. C. Tseng, D. B. Stolz, S. P. Wu, S. C. Watkins and L. Huang, Gene Ther., 1999, 6, 585–594 CrossRef CAS PubMed.
- S. P. Kasturi, K. Sachaphibulkij and K. Roy, Biomaterials, 2005, 26, 6375–6385 CrossRef CAS PubMed.
- S. Jain, P. U. Valvi, N. K. Swarnakar and K. Thanki, Mol. Pharmaceutics, 2012, 9, 2542–2553 CrossRef CAS PubMed.
- M. L. Forrest, N. Gabrielson and D. W. Pack, Biotechnol. Bioeng., 2005, 89, 416–423 CrossRef CAS PubMed.
- M. B. Roufai and P. Midoux, Bioconjugate Chem., 2001, 12, 92–99 CrossRef PubMed.
- P. R. Dash, M. L. Read, K. D. Fisher, K. A. Howard, M. Wolfert, D. Oupicky, V. Subr, J. Strohalm, K. Ulbrich and L. W. Seymour, J. Biol. Chem., 2000, 275, 3793–3802 CrossRef CAS PubMed.
- S. Li, M. A. Rizzo, S. Bhattacharya and L. Huang, Gene Ther., 1998, 5, 930–937 CAS.
- X. Sun, P. Ma, X. Cao, L. Ning, Y. Tian and C. Ren, Drug Delivery, 2009, 16, 357–362 CrossRef CAS PubMed.
- S. R. Datir, M. Das, R. P. Singh and S. Jain, Bioconjugate Chem., 2012, 23, 2201–2213 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |