
One-pot synthesis of micro/nano structured β-Bi2O3 with tunable morphology for highly efficient photocatalytic degradation of methylparaben under visible-light irradiation†
Xin Xiao*,
Ruiping Hu,
Shunheng Tu,
Chunxia Zheng,
Huan Zhong,
Xiaoxi Zuo and
Junmin Nan*
School of Chemistry and Environment, South China Normal University, Guangzhou Key Laboratory of Materials for Energy Conversion and Storage, Guangzhou 510006, P. R. China. E-mail: xiaox@scnu.edu.cn; jmnan@scnu.edu.cn; Fax: +86-20-39310187; Tel: +86-20-39310255
First published on 21st April 2015
Abstract
β-Bi2O3 micro/nanostructures with tunable morphologies were synthesized via a one-pot solvothermal–calcining route, and their photocatalytic activity toward degrading methylparaben (MeP, a widely used preservative with estrogenic activity) was evaluated under visible-light (λ ≥ 420 nm) irradiation. The formation process of β-Bi2O3 catalysts can be described as reduction of Bi3+ through a solvothermal reaction, followed by oxidization of metal Bi via calcination in air. During this process, the organic reductants (single or a mixture of ethylene glycol, D-fructose, and ascorbic acid) play important roles in determining the final morphologies and structures of the materials. Photocatalytic tests reveal that MeP can be effectively degraded and mineralized by using synthetic β-Bi2O3 catalysts, and the reaction rate constant of an optimum sample is more than 25 and 160 times faster than a commercial Bi2O3 and synthetic N-TiO2, respectively. The superior photocatalytic activity of the optimum product is ascribed to its pure beta phase with a narrower band gap, good absorption of visible light, more efficient separation of electrons and holes, relatively higher BET specific surface area, and three-dimensional architectures, which favor more surface active sites and easier mass and photoinduced charge transportations. In addition, the main reactive oxygen species and possible degradation intermediates were detected, and the results suggest that photogenerated holes and superoxide radicals are the predominant species in the photochemical oxidation process.
1. Introduction
Parabens, alkyl and aryl esters of para-hydroxybenzoic acid, are a group of preservatives widely used in cosmetics, pharmaceuticals, foodstuffs, and other health and personal care products.1 The human population is easily exposed to parabens from different sources since these chemicals have been used in over 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 formulations developed by various industries, resulting in an approximate global consumption of 8000 tons annually.2 In recent years, the commercial use of parabens has been continually growing, and as a consequence their residues are frequently detected in wastewater, surface water, groundwater, and drinking water,3,4 and even in human serum samples.5 Unfortunately, recent studies have shown that parabens are endocrine-disrupting compounds (EDCs) with estrogenic activity,6,7 and a possible relationship between breast cancer and the application of parabens has been speculated.8 As a result, parabens have been classified as emerging environmental contaminants by the U.S. Environmental Protection Agency.9 Since conventional wastewater treatment plants (WWTPs) cannot fully remove these antimicrobial preservatives,10 it is essential to develop effective methods to eliminate these compounds from parabens wastewater.
200 formulations developed by various industries, resulting in an approximate global consumption of 8000 tons annually.2 In recent years, the commercial use of parabens has been continually growing, and as a consequence their residues are frequently detected in wastewater, surface water, groundwater, and drinking water,3,4 and even in human serum samples.5 Unfortunately, recent studies have shown that parabens are endocrine-disrupting compounds (EDCs) with estrogenic activity,6,7 and a possible relationship between breast cancer and the application of parabens has been speculated.8 As a result, parabens have been classified as emerging environmental contaminants by the U.S. Environmental Protection Agency.9 Since conventional wastewater treatment plants (WWTPs) cannot fully remove these antimicrobial preservatives,10 it is essential to develop effective methods to eliminate these compounds from parabens wastewater.
To date, several methods have been reported in literatures for the treatment of parabens wastewater, including physical adsorption,11 biodegradation,12 photodegradation,13 photolysis of hydrogen peroxide (H2O2/UV),14 Fenton-like reactions,1 ozone oxidation,15 electrochemical processing,2 and photocatalytic degradation.9,16,17 Among them, the heterogeneous photocatalytic process seems to be an effective treatment technique due to it has high degradation and mineralization efficiency, low toxicity, low cost, and ability to perform under ambient conditions.18 However, although the photocatalytic decomposition of parabens using TiO29,16,17,19 and ZnO20–22 photocatalysts has been an active field in recent years, to the best of our knowledge, the photocatalytic degradation of parabens under visible-light irradiation has not been explored, and the mechanism governing the visible-light-induced photocatalytic degradation of parabens still remains unknown.
As the simplest Bi-based oxide, Bi2O3 has captured increased attention due to its excellent properties, including a wide energy gap range (2–3.96 eV), high refractive index, high dielectric permittivity, marked photoconductivity and photoluminescence.23,24 As a result, it has been widely employed in gas sensors for NO and NO2 detection, optoelectronics devices, optical coatings, and ceramic glass manufacturing.24 Bi2O3 has five polymorphic forms: monoclinic α, tetragonal β, body-centered cubic γ, cubic δ, and triclinic ω.25 Previous theoretical studies have revealed that in Bi2O3, the top of valence bands (VBs) is mainly comprised of O 2p and Bi 6s orbitals, while the bottom of its conduction bands (CBs) is dominantly constructed by Bi 6p orbital.26–29 However, although the CBs of Bi2O3 polymorphs are similar, their structures of VBs are various, and the VB top of β-Bi2O3 shifts to a lower potential, resulting in its smaller band gap.26 This diversity of Bi2O3 polymorphs in band structures could result from their different crystal and electronic structures, in which the Bi–O bond interaction plays a key role. For β-Bi2O3, the maximal binding is observed for two nearest O atoms instead of strong Bi–O interactions, resulting a stronger hybridization of the O 2p and Bi 6s, 6p orbitals in its VBs, thus the wide band of the O 2p hybridized states is split into subbands to gain a narrower band gap.30 In addition, the band structures analysis also shown that β-Bi2O3 exhibits a more dispersive band structures which is suitable for the transfer of the photoinduced electrons and holes, and the tunnels in its distinctive crystal structure can further provide the channels for the photogenerated electrons and holes transfer to prevent the excessive recombination of them.27 Consequently, the β-Bi2O3 has been recommended that as a promising visible-light-driven photocatalyst thanks to its relatively small band gap, strong absorption of light over the UV-visible range, better separation of photogenerated carriers, high oxidation power, and non-toxic nature.26
Until now, the general route for synthesizing β-Bi2O3 has been the thermal decomposition of bismuth-oxide compounds, such as bismuth subcarbonate,31 bismuth oxalates,32 [Bi38O45(OMc)24(DMSO)9]·2DMSO·7H2O,33 and (H2O)0.75Bi2(CH3COO)(NO3)1.12.34 Recently, we have found that nano β-Bi2O3 can be easily prepared by direct calcination of nanometallic bismuth at a relatively low temperature in air.35 It is well known that the photocatalytic activities of photocatalysts are greatly influenced by their structures and morphologies. For example, three-dimensional (3D) micro/nanoarchitectures made-up from nanosized building blocks may provide advantages, such as high surface area, anti-aggregation ability, and abundant transport paths for the degradation reaction, resulting in their enhanced photocatalytic performances.36 Therefore, our previous finding may disclose a new way to obtain β-Bi2O3 with desired structures and morphologies by just controlling the properties of the metallic bismuth precursors, which offers an opportunity to improve the photocatalytic efficiency of nano β-Bi2O3 for efficient contaminants removal.
In this study, micro/nanostructured β-Bi2O3 photocatalysts with tunable morphologies were synthesized through a facile one-pot solvothermal–calcining technique, and methylparaben (MeP), one of the most commonly used parabens, was chosen as the target pollutant. The morphology, structure, photoabsorption, nitrogen adsorption properties, photocurrent responses, and formation process of as-synthesized β-Bi2O3 samples were systematically characterized and analyzed. Then, the photocatalytic activity and mineralization efficiency of the as-prepared samples toward the photocatalytic degradation of MeP under visible-light irradiation were evaluated. In addition, the degradation intermediates of MeP and photogenerated reactive species in the system were identified, and a possible photocatalytic degradation mechanism was proposed based on the experimental results.
2. Experimental
2.1. Materials and methods
Methylparaben (purity: 99.99%) and nitroblue tetrazolium (NBT) were purchased from J&K Chemical Ltd. Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O) was obtained from Tianjin Kermel Chemical Reagent Co., Ltd. Ethylene glycol (EG) was provided by Chinasun Specialty Products Co., Ltd. D-Fructose was purchased from Shanghai Bio Science & Technology Co., Ltd. Ascorbic acid (AA) was supplied by Sinopharm Chemical Reagent Co., Ltd. A commercially available β-Bi2O3 was purchased from Aladdin Reagent Co., Ltd. All other chemicals were of analytical grade and used without further purification.In a typical synthesis procedure, 0.728 g Bi(NO3)3·5H2O was dissolved completely in 35 mL of EG and maintained stirring at room temperature. Then, 0.540 g D-fructose and 0.100 g AA were sequentially added to the solution. The mixture was then transferred into a Teflon-lined stainless-steel autoclave and heated at 180 °C for 4 h in an oven. After completing the solvothermal reaction, the precipitates were harvested by centrifugation, washed with distilled water and ethanol for three times to remove any ionic residue, and dried in an oven at 60 °C. Finally, the product obtained was calcined in air at 290 °C for 1 h in a muffle furnace. The sample synthesized with D-fructose and AA was denoted as S1, that synthesized with AA alone was denoted as S2, and that synthesized with D-fructose alone was denoted as S3, while the sample synthesized without the addition of D-fructose and AA was denoted as S4.
For comparison, a nitrogen-doped TiO2 sample was obtained by a solvothermal method used ethylenediamine as the nitrogen source according to the method of Yang et al.37
2.2. Catalyst characterization
The crystalline structures and phase compositions of the as-prepared samples were determined by powder X-ray diffraction (XRD) on a Bruker D8 Advance X-ray diffractometer (Bruker AXS, Germany) with Cu Kα radiation. The surface morphologies of the samples were studied using a field-emission scanning electron microscope (FE-SEM, S-4800, Hitachi). UV-Vis diffuse reflection spectra (DRS) were recorded on a UV-Vis spectrophotometer (UV-3010, Hitachi, Japan) applying an integrating sphere with BaSO4 as a reference, and were converted to absorption spectra by the Kubelka–Munk method. The specific surface areas and average pore diameter of the samples were evaluated using nitrogen adsorption–desorption isotherms according to the Brunauer–Emmett–Teller method (BET, ASAP 2020, Micromeritics, USA). A desorption isotherm was applied to calculate the pore size distribution using the Barrett–Joyner–Halenda (BJH) method. Photocurrent measurements were performed on a CHI 660C electrochemical station (Chenhua Instruments Co. Shanghai, China) in a standard three-electrode configuration containing the as-prepared samples as the working electrodes, a platinum plate as the counter electrode, and a commercial Ag/AgCl electrode as the reference electrode, with 0.5 mol L−1 Na2SO4 aqueous solution as the electrolyte. The working electrodes were fabricated by dispersing a certain amount of samples in ethylcellulose–ethanol solution, following deposited the suspension onto the surface of fluorine-doped transparent conductive oxide (FTO) glass with a defined area controlled by plastic insulation, and then tried in an oven at 60 °C to yield film electrodes. A 300 W xenon lamp (PLS-SXE300/300UV, Beijing Trusttech Co. Ltd., China) assembled with a UV light cut-off filter (λ > 420 nm) was used as the visible-light source. The Mott–Schottky experiments were conducted to evaluate the band structure of as-synthesized samples with an electrochemistry workstation (Princeton PARSTAT 2273, USA) using a working electrode (an appropriate amount of the sample suspension on a conductive indium tin oxide glass), a platinum plate as counter electrode, and an Ag/AgCl electrode as reference electrode. A 0.5 mol L−1 Na2SO4 aqueous solution was used as the electrolyte. The potential range was −0.8 to +0.6 V at a constant frequency of 1000 Hz. Electron paramagnetic resonance (EPR) spectrometry (JES FA-200, JEOL, Japan) was used to record signals of ˙OH and ˙O2− by the reaction with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). The settings for EPR spectrometer were center field of 323 mT, sweep width of 5 mT, microwave frequency of 9055 MHz, power of 0.998 mW, modulation frequency of 100 kHz.2.3. Photocatalytic activity measurements
Photocatalytic activity tests were conducted in a photochemical reactor (XPA-VII, Xujiang, Nanjing, China) equipped with a 1000 W Xe lamp together with a 420 nm cut-off filter as the visible-light source. The experiments were performed as follow: 50 mg of as-synthesized photocatalyst was added into a 50 mL of MeP solution (10 mg L−1). Prior to irradiation, the suspension was stirred for 1 h in the dark to ensure the adsorption–desorption equilibrium between photocatalyst powders and MeP. During the photocatalytic process, about 3 mL of the suspension was collected at a specified time; subsequently, the solids were separated from the solution using a 0.45 μm nitrocellulose filter, and the filtrate was then analyzed by recording the absorbance at the characteristic band of 255 nm using a UV-Vis spectroscopy (UV-1800, Shimadzu, Japan) to determine the MeP concentrations. The NBT, which exhibits a maximum absorption at 259 nm and can be specifically reduced by superoxide radicals to produce insoluble purple formazan, was chosen as a molecular probe to clarify the ˙O2− generating from as-prepared photocatalysts under visible-light irradiation. Experiments were carried out using a procedure same as photocatalytic reactions but simply replaced MeP by 25 mg L−1 NBT solution. The total organic carbon (TOC) content was determined using an automatic total organic carbon analyzer (TOC-V, Shimadzu, Japan).2.4. Analysis of the photogeneration intermediates
After the photocatalytic reaction for certain time, sample solutions were collected, separated, and extracted three times with an ethyl acetate/n-hexane mixture (2:1, v/v). Afterwards, the organic layers were combined and concentrated to approximately 1 mL under a gentle stream of high-purity nitrogen. The intermediates were investigated by gas chromatography-mass spectrometry (GC-MS, DSQ/Trace GC Ultra, Thermo Fisher Scientific, Dreieich, Germany) on a DB 5-MS column (30 m × 0.25 mm). The GC column temperature was programmed from 50 °C to 250 °C at 5 °C min−1 and maintained at 250 °C for 5 min. The injector and transfer line temperatures were 230 °C. Helium gas was used as the carrier gas at a constant flow rate of 1.2 mL min−1. The ion MS source temperature was 250 °C, and ionization was performed by electron ionization at 70 eV.
2.5. Calculation of frontier electron density
Quantum calculations using Gaussian 09 were carried out to get the frontier electronic density (FED) of MeP. Hartree–Fock (HF) theory was used for the full optimization of geometries, and B3LYP/6-311++G(d,p) was used as the basis set of moderate size and accuracy. All optimizations were performed without any symmetry restrictions using the default convergence criteria in the programs and were followed by harmonic frequency analyses to ensure that the optimized conformation was the true global minimum.3. Results and discussion
3.1. Characterization of catalysts
The as-synthesized samples before and after calcination were characterized by XRD to explore their chemical compositions and phase structures. As shown in Fig. 1, all diffraction peaks of the samples before calcination can be unambiguously indexed as rhombohedral bismuth (JCPDS no. 85-1329, Fig. 1A), while the samples were found to be tetragonal Bi2O3 (JCPDS no. 78-1793) after calcination at 290 °C (Fig. 1B). Moreover, it can be seen that the crystallinity of sample S4 before the heat treatment was poor, and some impurity peaks appeared after the calcination, which was recognized as the existence of a small amount of monoclinic α-Bi2O3 (JCPDS no. 71-2274). The analysis can be further verified by calcining the sample S4 at a slightly higher temperature (Fig. S1, ESI†). The results of XRD reveal that under the current synthesis route, metallic Bi were obtained by the solvothermal reaction, then it could subsequently be converted to β-Bi2O3 after thermal treatment in air.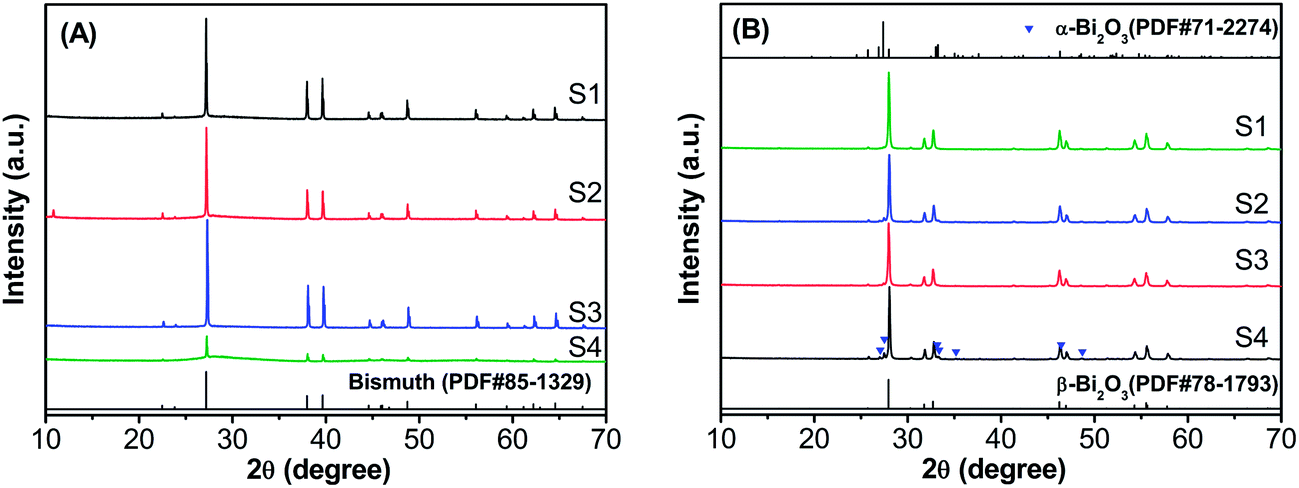 | ||
| Fig. 1 XRD patterns of the as-synthesized samples (A) before and (B) after calcination. | ||
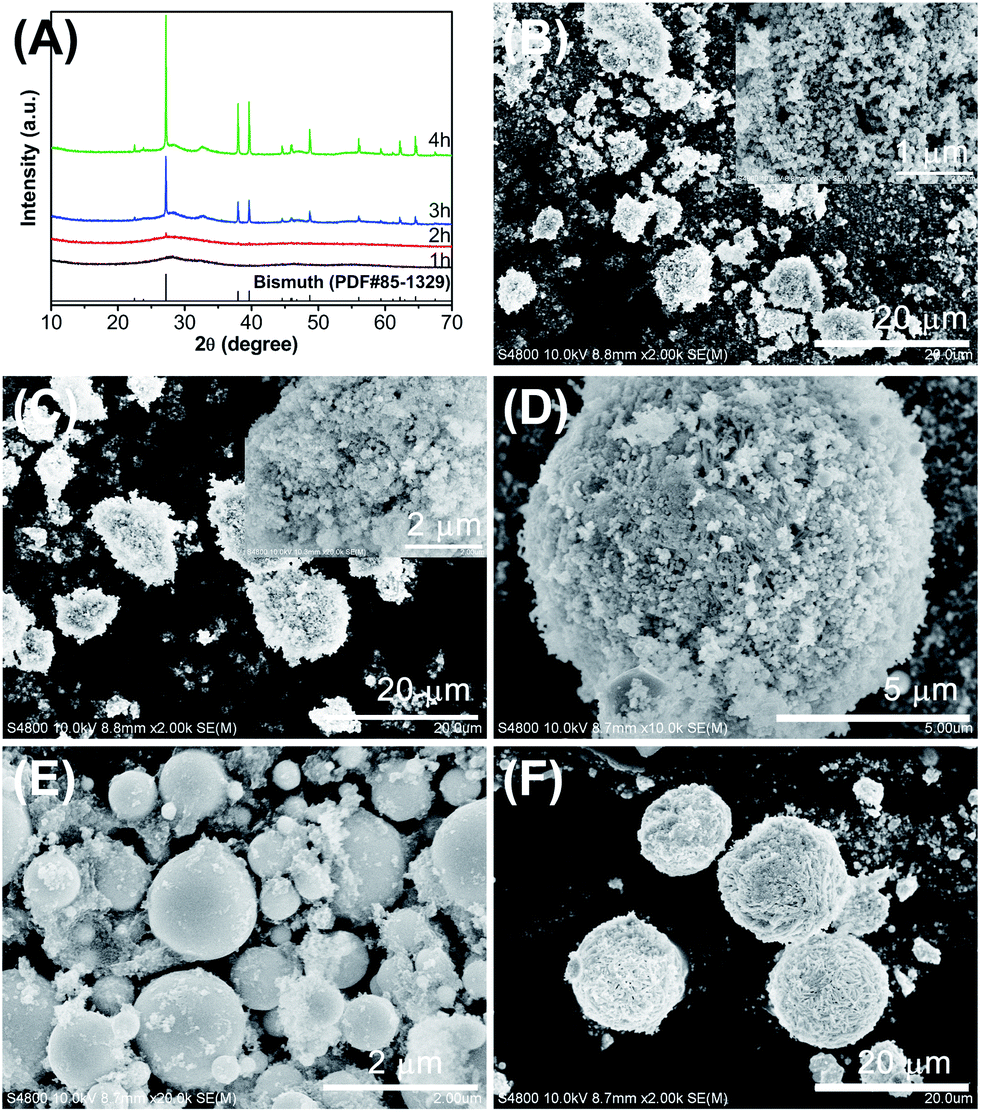
The morphologies and surface structures of samples S1–S4 were investigated by SEM. As shown in Fig. 2, the samples overall exhibited agglomerated, irregular sphere-like morphologies with different grain sizes. Sample S3, S1, and S2 showed apparent diameters of approximately 1 μm, 5 μm, and 10 μm, respectively, whereas sample S4 showed a greater diameter of approximately 20 μm. In addition, the samples were observed to be very different in microstructures. Sample S1 (Fig. 2A) exhibits numerous nanosheets with a thickness of less than 20 nm, which connect to one another and fabricate three-dimensional microparticles. Sample S2 (Fig. 2B) consists of numerous nanoparticles aggregated to form spherical structures. Sample S3 (Fig. 2C) has a large number of individual microspheres with a smooth surface. And sample S4 (Fig. 2D) also presents a three-dimensional microsphere-like morphology; however, these microspheres are composed of many sub-micron sheets with a thickness about 200 nm. The results suggest that β-Bi2O3 with an adjustable morphology can be synthesized by an one-pot process with introduce of different additives. It is well known that there is a close relationship between the size, shape, microstructure, and properties of materials. Therefore, catalysts with adjustable microstructures are expected to be different levels of photocatalytic activity.
 | ||
| Fig. 2 SEM images of the as-synthesized samples: (A–D) samples S1–S4. The insets show a high-magnification SEM image of each corresponding sample. | ||
A series of time-dependent experiments has been carried out to explore the possible formation mechanism of the as-synthesized samples with different morphologies, and the obtained products were studied by XRD and SEM. The XRD patterns of the precursor of sample S1 obtained after solvothermal reaction for 1, 2, 3, and 4 h are shown in Fig. 3A and S2 (ESI†). The patterns clearly show that the typical diffraction peaks of metallic bismuth (JCPDS no. 85-1329) simultaneously increased with the reaction time. These results imply that a reduction reaction occurs during the solvothermal process, through which Bi3+ is gradually converted to Bi0. It is noteworthy that in addition to the peaks of bismuth, some extra diffraction peaks with lower intensity can be also observed in Fig. 3A and S2,† including 28.4, 32.8, and 47.1 degrees. Then these peaks can be identified as BiO2−x (JCPDS no. 47-1057), yet the exact formation mechanism of this species needs further exploration. In addition, the products obtained after a reaction for 1 h (Fig. 3B) or 4 h (Fig. 3C) are not much different in morphology and exhibit a little change before (Fig. 3C–F) and after calcination (Fig. 2A–D), suggesting that the shape of the final products is largely determined by their early aggregated form in the solvothermal reaction.
 | ||
| Fig. 3 (A) XRD patterns of the sample S1 precursor after solvothermal reaction for 1, 2, 3, and 4 h, respectively; (B) SEM images of the sample S1 precursor after solvothermal reaction for 1 h; (C–F) SEM images of the sample S1–S4 precursors after solvothermal reaction for 4 h, respectively. | ||
From above analysis based on the XRD and SEM, the formation of β-Bi2O3 with different morphologies may be described by a reduction process by solvothermal reaction and oxidation of Bi metal via heat treatment, which similar to that reported in our previous study.35 During this process, EG may act as a complexing agent reacted with Bi3+ to form glycolate complexes to avoid the hydrolysis of Bi(NO3)3.38 Within a solvothermal process, EG (a polyol that can produce acetaldehyde at high temperature39) alone or mixed with D-fructose (a ketone-bearing sugar that can readily transform into reducing sugar because ketoenol tautomerism40) and AA (based on the reducing properties of its enediol group41) act as reducing agents to obtain metallic bismuth, and the reaction conditions (reductants and reaction temperature) critically affect the morphologies and structures of the Bi precursors. After subsequent annealing in an oxygen atmosphere, the metallic Bi reacts with O2 to form bismuth oxides. At a comparatively low calcination temperature (290 °C), a metastable phase of bismuth oxide (β-Bi2O3) can be achieved. The low crystallinity (before calcination) and the impurity XRD peaks (after calcination) of sample S4 may be attributed to the lower reducing power of EG alone.
It is known that the optical absorption of a semiconductor is one of key factors in determining its photocatalytic activity. The optical absorption of as-prepared samples S1–S4 was measured using diffuse reflectance spectra (DRS) and compared in Fig. 4. It reveals the absorption edge of sample S1, S2, and S3 are about 542, 544, and 548 nm, respectively. Nevertheless, sample S4 presented two absorption bands, and the absorption edge of them can be estimated to be 466 and 541 nm, respectively, which reflects the multi-phase nature of the sample S4 (Fig. S1†). The results is consistent with their light yellow color (inset in Fig. 4) and suggests they could efficiently absorb visible light. The band gap energies (Eg) of the samples were calculated using the equation (eqn (1)):42
| α(hν) = A(hν − Eg)n/2 | (1) |
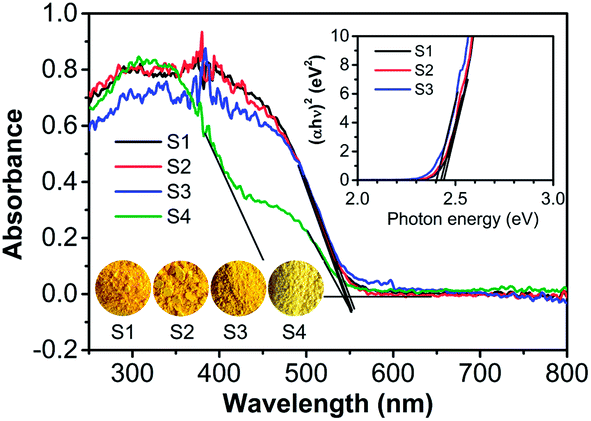 | ||
| Fig. 4 UV-Vis diffuse reflection spectra (DRS) of samples S1–S4. The upper inset shows plots of (αhν)2 vs. photon energy (hν), and the lower inset shows photographs of the samples. | ||
N2 adsorption–desorption isotherms were conducted to investigate the surface area and pore structures of as-obtained samples S1–S4. As shown in Fig. 5, all the isotherms are belong to type IV with an H3 hysteresis loop as defined by IUPAC, which is characteristic of mesoporous materials.44 The BET surface areas of the samples S1–S4 were then estimated using N2 isotherms to be 5.18, 4.01, 3.66, and 3.88 m2 g−1, and the corresponding pore size distributions were calculated using the BJH method to be 49.2, 46.9, 45.9, and 43.6 nm (inset in Fig. 5), respectively. The mesoporous structure with wide pore size distribution may arise from the inter-nanosheet/particle spaces within the samples. The relatively small specific surface area may be ascribed to the particles aggregation that occurred in these samples after the calcination.
 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms and the corresponding pore size distribution curve (inset) of the as-synthesized samples S1–S4. | ||
It is generally believed that the photocatalytic activity of a catalyst depends on its ability to generate and separate photo-induced charge carriers. Fig. 6 showed the transient photocurrent responses via on–off cycles of each sample electrode under visible-light irradiation, which may be directly correlated with the separation and recombination efficiency of photogenerated carriers. It can be found that the sample S1 exhibited the highest photoelectric current response among these samples, which indicates its higher ability of charge generate and separation. The better separation and transfer of photogenerated charge carriers of sample S1 may be associated with its high pure β-phase and unique 3D architecture composed of numerous ultrathin 2D nanosheets which allow for faster electron transport.
 | ||
| Fig. 6 Photocurrent transient responses of the as-synthesized samples S1–S4. | ||
3.2. Photocatalytic degradation of methylparaben
The photocatalytic activities of the as-fabricated samples S1–S4 were evaluated through the degradation of MeP solution and compared with that of direct photolysis (Blank), a N-doped TiO2, and a commercially available β-Bi2O3 sample under visible-light irradiation. Fig. 7A shows the degradation efficiencies of the MeP solution by the spots of C/C0 vs. irradiation time, where C is the concentration of the remaining of MeP, while C0 is the initial concentration of MeP solution after adsorption equilibrium. It is obvious that MeP was hardly degraded by direct visible-light irradiation (Blank). When submitted to identical irradiation conditions for 60 min, the N-doped TiO2 sample showed only 4.2% degradation and the commercial β-Bi2O3 sample presented a degradation of 28.2%. The as-synthesized samples S1–S4, on the other hand, exhibited much higher degradation efficiencies of 99.9%, 99.6%, 98.9%, and 48.2%, respectively, demonstrating that the as-synthesized samples S1–S3 possessed excellent visible-light-induced photocatalytic performances. And among them, sample S1 revealed the best activity. To estimate the influence of samples' surface area on the MeP removal. A profiles of MeP elimination by combination of photodegradation and dark-adsorption was presented in Fig. S3 (ESI†). The results showed that the adsorption capacity of the samples consistent with the trend of their corresponding specific surface area, and the sample S1 has a maximum adsorption of 19% before photodegradation, which is benefit its photocatalytic performance. | ||
| Fig. 7 (A) Photocatalytic degradation kinetics of MeP over the as-synthesized samples S1–S4, commercial Bi2O3 (C-Bi2O3), N-doped TiO2 (N-TiO2), and a blank sample (without catalyst) under visible-light irradiation. (B) Linear plots of ln(C0/Ct) versus degradation time. | ||
Furthermore, the kinetics of MeP photodegradation over the as-synthesized samples, commercial β-Bi2O3, and N-TiO2 can be further investigated by fitting the experimental data to the following pseudo-first-order kinetics equation (eqn (2)):45
 | (2) |
As is well known, the photocatalytic efficiency of a catalyst is governed by various factors, such as composition and phase structure, optical absorption properties (band gap energy), the oxidation potential of photogenerated holes, the separation efficiency of photogenerated electrons and holes pairs, surface area, and so on. Compared with other samples, sample S1 had a high pure β phase, good photoabsorption from UV to visible-light, fast photogenerated charge separation and interfacial charge transfer, higher specific surface area with mesoporous feature, and a unique three-dimensional structure composed of numerous ultrathin 2D nanosheets, resulting in its superior photocatalytic performance for the degradation of MeP under visible-light irradiation. Then, the sample S1 was used as the optimum catalyst in the following experiments.
The removal of total organic carbon (TOC) was chosen as a mineralization index for the decomposition of MeP in the presence of the sample S1 as catalyst during the photocatalytic process. The time dependency of the TOC concentration is shown in Fig. 8. It can be seen that about 66.8% TOC was decomposed to inorganic compounds after visible-light irradiation for 60 min, indicating that mineralization reaction is very fast. In addition, compared with the TOC degradation of ∼35% from the same initial concentration of MeP for an identical irradiation time using nano-TiO2 as a photocatalyst and UV light as a light source,9 the TOC removal in current work demonstrates that MeP can be effectively mineralized using β-Bi2O3 as photocatalyst under visible light irradiation.
 | ||
| Fig. 8 TOC removal efficiency of MeP using the as-synthesized β-Bi2O3 (sample S1) under visible-light irradiation. | ||
To examination the stability and reusability of the as-synthesized sample, the sample S1 was reused for three times (once a day for three days) under the same conditions, and compared with the commercial β-Bi2O3. As displayed in Fig. S4 (ESI†), the results showed that the recyclability of commercial β-Bi2O3 sample is very poor, which may be ascribed to its structural instability during photocatalytic process,46 while the sample S1 demonstration a good stability during three cycles, which is important for its practical application.
3.3. Identification of intermediates and photocatalytic mechanism
To explore the degradation intermediates of MeP, during the photocatalytic process, the reaction solution was separated, extracted, concentrated, and then examined with GC/MS technique. As shown in Fig. 9, all components with retention times (RT) less than 12 min, MeP were eluted at a RT of 9.23 min and disappeared rapidly after the photoreaction, which is in accordance with the UV-Vis spectroscopy data. During the degradation process, four intermediates including tartaric acid (RT = 3.81), phenol (RT = 4.71), 6-hydroxyhexan-2-one (RT = 5.18), and 4-methylpentanoic acid (RT = 5.98) were identified. Previous reports on the degradation of MeP using advanced oxidation processes (AOPs) have suggested that the degradation pathways proceed mainly through the hydroxylation of MeP due to the presence of reactive hydroxyl radicals (˙OH).1,2,9,15 However, in this visible-light-induced β-Bi2O3 photodegradation system, no hydroxylated compounds were identified, which implies it has a different mechanism of photocatalytic oxidation of MeP.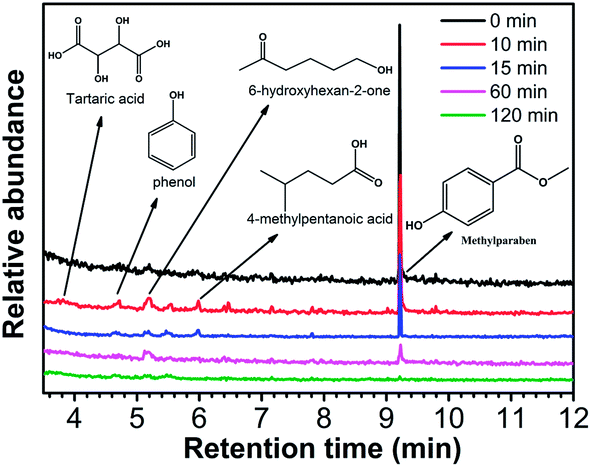 | ||
| Fig. 9 GC-MS chromatograms of MeP degradation over an as-synthesized β-Bi2O3 sample (sample S1) under visible-light irradiation. | ||
To investigate the role of the major reactive species on the photodegradation reaction, the surface radicals and holes trapping experiments were performed by adding individual scavengers to the photodegradation system with the method similar to the former photocatalytic activity experiments. Among them, isopropanol (IPA) was used to scavenge ˙OH, sodium oxalate was used to scavenge photogenerated h+, KBrO3 was used to scavenge photogenerated e−, 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (TEMPOL) was used to scavenge ˙O2−, and N2 was used to remove oxygen that dissolved in the solution.47 As illustrated in Fig. 10A, the photocatalytic degradation efficiency of MeP has no obvious decrease after adding isopropanol as a sacrificial donor. However, recognizable inhibitions of the photocatalytic degradation were occurred when performed in the presence of KBrO3, TEMPOL, sodium oxalate, or N2, suggesting the significance of superoxide anion radicals and photogenerated holes in this process. This conjecture may be further confirmed by the EPR method (Fig. 10B), which reveals strong DMPO–˙O2−-adduct signals after visible-light irradiation.
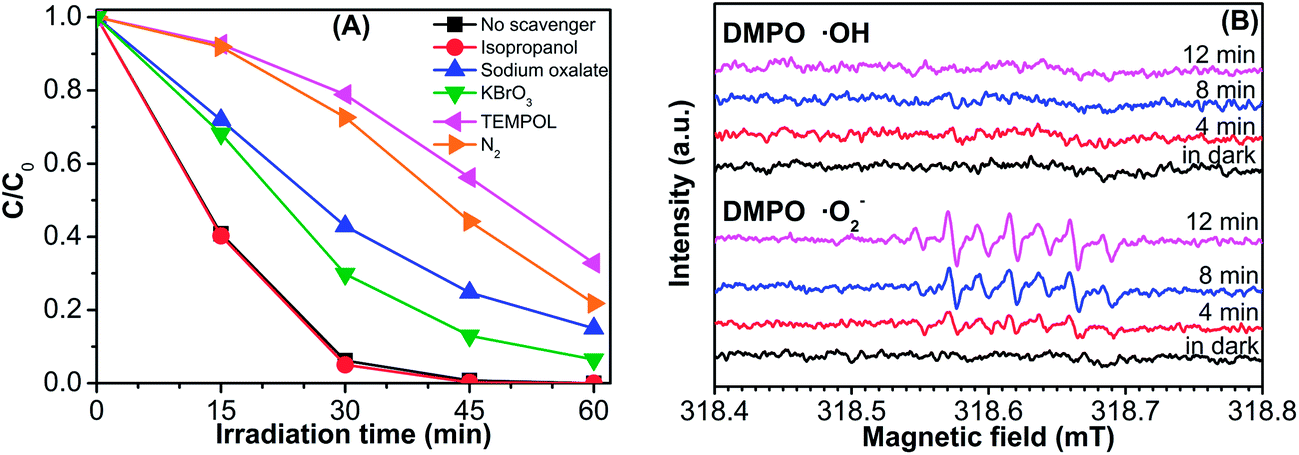 | ||
| Fig. 10 (A) Photodegradation of MeP over an as-synthesized β-Bi2O3 sample in the presence of different scavengers: no scavenger, isopropanol, sodium oxalate, KBrO3, TEMPOL, and N2. (B) DMPO spin-trapping EPR spectra for DMPO–˙OH and DMPO–˙O2− under visible-light irradiation with a β-Bi2O3 sample. | ||
It is generally believed that the conduction band (CB) minimum of Bi2O3 polymorphs are close to NHE.29,48 In order to confirm this inference, the flatband potentials of as-prepared samples S1 and S2 were determined from Mott–Schottky measurements. As shown in Fig. S5 (ESI†), the flatband potentials of sample S1 and S2 are estimated to be −0.28 V and −0.26 V vs. Ag/AgCl (−0.09 V and −0.07 V vs. NHE), respectively. Thus, the low conduction band potential of β-Bi2O3 seems cannot reaction of O2 to produce ˙O2− (−0.33 V vs. NHE). However, due to the valence band potential of β-Bi2O3 is about 2.3 V vs. NHE according to the above analysis. Under λ ≥ 420 nm (E ≤ 2.95 eV) light irradiation, some of electrons can be excited to be more negative potentials to reaction with oxygen to generate superoxide radicals. To further confirm this hypothesis, the sample S1 calcined at 290 °C (β-Bi2O3) and 500 °C (α-Bi2O3) were used to photodegrade NBT to determine the amount of ˙O2− generating.49 As can be observed from the Fig. S5 (ESI†) that after 120 min of visible-light irradiation, about 22% of NBT was degraded over sample S1 (calcined at 290 °C), confirming the generation of photoelectrons and ˙O2−. For comparison, the NBT hardly be degraded by α-Bi2O3 sample (calcined at 500 °C) after identical process, which is related to its more positive valence band potential (∼2.8 V vs. NHE).
Consequently, the photocatalytic degradation of MeP using the as-prepared β-Bi2O3 photocatalyst under visible-light may be dominated by directed holes and superoxide anion radicals oxidation rather than reaction with ˙OH. This phenomenon may be explained by the standard redox potential of Bi5+/Bi3+ (E0 = 1.59 V at pH 0) is more negative than that of ˙OH/OH− (E0 = 1.99 V at pH 0). Therefore, the photogenerated holes on the surface of catalysts could not oxidize H2O to yield ˙OH.50
A theoretical calculation of the frontier electron density (FED) of MeP (Fig. 11) was employed to promote understand the photodegradation intermediates. According to frontier orbital theory, electrophilic, nucleophilic, and radical reactions may occur at different atomic of a molecular: the positions with higher values of FEDHOMO2 + FEDLUMO2 are more susceptible to a radical attack,51 while positions with higher value of 2FEDHOMO2 are more easily subject to an electron extraction.52 The calculated results are summarized in Table 1, which clearly suggests that the highest 2FEDHOMO2 value of MeP occurs at C4. Consequently, C4 may be the first site from oxidized by photogenerated holes. The formed phenols can be further oxidized, resulting in the breakdown of aromatic structures subsequently production of carboxylate acid and evolution of CO2. Eventually, the MeP is mineralized into inorganic molecules through the photodegradation process.
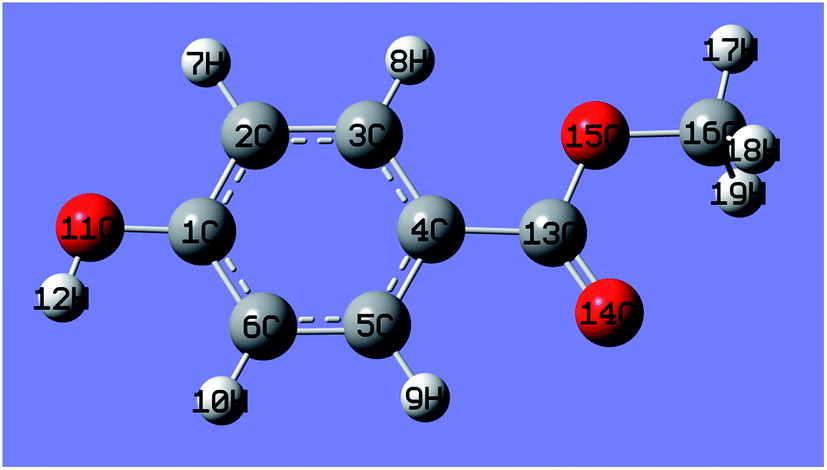 | ||
| Fig. 11 The molecular structure of MeP with atoms labeled. | ||
| Atom label | 2FEDHOMO2 | FEDHOMO2 + FEDLUMO2 |
|---|---|---|
| C1 | 0.314 | 0.695 |
| C2 | 0.133 | 1.149 |
| C3 | 0.087 | 0.703 |
| C4 | 0.339 | 0.881 |
| C5 | 0.246 | 2.190 |
| C6 | 0.102 | 1.454 |
| O11 | 0.093 | 0.063 |
| C13 | 0.190 | 0.316 |
| O14 | 0.077 | 0.158 |
| O15 | 0.012 | 0.056 |
| C16 | 0.015 | 0.063 |
4. Conclusions
In this work, we have synthesized micro/nano β-Bi2O3 materials with tunable morphology via a facile one-pot solvothermal–calcining process using EG, AA, and D-fructose as mild organic reducing agent. XRD and SEM observation of the samples before and after the calcination indicated that the β-Bi2O3 were developed by reduction of Bi ions to form metallic bismuth precursors with different morphologies by a solvothermal reaction. Thereinto, EG acts as a solvent and complexing agent, EG, AA, and D-fructose serve as reducing agents as well as structure-directing agents, and followed by oxidization of metal Bi after the calcination in air, in which O2 serves as an oxidizing agent. The as-synthesized β-Bi2O3 samples exhibited effective photoabsorption from UV to visible-light regions and high photogenerated charge separation ability, resulting in their excellent photocatalytic properties in the degradation of MeP under visible-light irradiation. Besides, photogenerated holes and superoxide radicals were confirmed to be the governing reactive oxygen species in the photodegradation and mineralization of MeP.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21477040), the Natural Science Foundation of Guangdong Province of China (no. S2012040007074), and the Scientific Research Foundation of Graduate School of South China Normal University (2014ssxm29).References
- X. Feng, Y. Chen, Y. Fang, X. Wang, Z. Wang, T. Tao and Y. Zuo, Sci. Total Environ., 2014, 472, 130 CrossRef CAS PubMed.
- J. R. Steter, R. S. Rocha, D. Dionísio, M. R. Lanza and A. J. Motheo, Electrochim. Acta, 2014, 117, 127 CrossRef CAS PubMed.
- E. Eriksson, H. R. Andersen and A. Ledin, J. Hazard. Mater., 2008, 156, 240 CrossRef CAS PubMed.
- B. R. Ramaswamy, J. Kim, T. Isobe, K. Chang, A. Amano, T. W. Miller, F. P. Siringan and S. Tanabe, J. Hazard. Mater., 2011, 192, 1739 CrossRef CAS PubMed.
- X. Ye, X. Zhou, L. Wong and A. M. Calafat, Environ. Sci. Technol., 2012, 46, 12664 CrossRef CAS PubMed.
- H. Yamamoto, I. Tamura, Y. Hirata, J. Kato, K. Kagota, S. Katsuki, A. Yamamoto, Y. Kagami and N. Tatarazako, Sci. Total Environ., 2011, 410–411, 102 CrossRef CAS PubMed.
- M. G. Soni, S. L. Taylor, N. A. Greenberg and G. A. Burdock, Food Chem. Toxicol., 2002, 40, 1335 CrossRef CAS.
- P. D. Darbre and A. K. Charles, Anticancer Res., 2010, 30, 815 CAS.
- Y. Lin, C. Ferronato, N. Deng, F. Wu and J. Chovelon, Appl. Catal., B, 2009, 88, 32 CrossRef CAS PubMed.
- B. Albero, R. A. Perez, C. Sanchez-Brunete and J. L. Tadeo, J. Hazard. Mater., 2012, 239–240, 48 CrossRef CAS PubMed.
- Y. P. Chin, S. Mohamad and M. R. Bin Abas, Int. J. Mol. Sci., 2010, 11, 3459 CrossRef CAS PubMed.
- A. Amin, S. Chauhan, M. Dare and A. K. Bansal, Eur. J. Pharm. Biopharm., 2010, 75, 206 CrossRef CAS PubMed.
- D. Bledzka, D. Gryglik and J. S. Miller, J. Photochem. Photobiol., A, 2009, 203, 131 CrossRef CAS PubMed.
- D. Bledzka, J. S. Miller and S. Ledakowicz, Ozone: Sci. Eng., 2012, 34, 354 CrossRef CAS.
- K. S. Tay, N. Abd Rahman and M. R. Bin Abas, Chemosphere, 2010, 81, 1446 CrossRef CAS PubMed.
- Y. Lin, C. Ferronato, N. Deng and J. Chovelon, Appl. Catal., B, 2011, 104, 353 CrossRef CAS PubMed.
- H. Fang, Y. Gao, G. Li, J. An, P. Wong, H. Fu, S. Yao, X. Nie and T. An, Environ. Sci. Technol., 2013, 47, 2704 CrossRef CAS PubMed.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- T. Velegraki, E. Hapeshi, D. Fatta-Kassinos and I. Poulios, Appl. Catal., B, 2015 DOI:10.1016/j.apcatb.2014.11.022.
- S. Lam, J. Sin, A. Z. Abdullah and A. R. Mohamed, Ceram. Int., 2013, 39, 2343 CrossRef CAS PubMed.
- S. Lam, J. Sin, A. Zuhairi Abdullah and M. Abdul Rahman, Mater. Lett., 2013, 93, 423 CrossRef CAS PubMed.
- J. Sin, S. Lam, K. Lee and A. R. Mohamed, Mater. Sci. Semicond. Process., 2013, 16, 1542 CrossRef CAS PubMed.
- L. Zhou, W. Wang, H. Xu, S. Sun and M. Shang, Chem.–Eur. J., 2009, 15, 1776 CrossRef CAS PubMed.
- F. Zheng, G. Li, Y. Ou, Z. Wang, C. Su and Y. Tong, Chem. Commun., 2010, 46, 5021 RSC.
- H. Lu, S. Wang, L. Zhao, B. Dong, Z. Xu and J. Li, RSC Adv., 2012, 2, 3374 RSC.
- H. Cheng, B. Huang, J. Lu, Z. Wang, B. Xu, X. Qin, X. Zhang and Y. Dai, Phys. Chem. Chem. Phys., 2010, 12, 15468 RSC.
- F. Wang, K. Cao, Q. Zhang, X. Gong and Y. Zhou, J. Mol. Model., 2014, 20, 1 CAS.
- H. Jiang, P. Li, J. Ye and J. Lin, J. Mater. Chem. A, 2015, 3, 5119 CAS.
- J. Zhang, W. Dang, X. Yan, M. Li, H. Gao and Z. Ao, Phys. Chem. Chem. Phys., 2014, 16, 23476 RSC.
- V. P. Zhukov, V. M. Zhukovskii, V. M. Zainullina and N. I. Medvedeva, J. Struct. Chem., 1999, 40, 831 CrossRef CAS.
- G. Zhu, J. Lian, M. Hojamberdiev and W. Que, J. Cluster Sci., 2013, 24, 829 CrossRef CAS.
- O. Monnereau, L. Tortet, P. Llewellyn, F. Rouquerol and G. Vacquier, Solid State Ionics, 2003, 157, 163 CrossRef CAS.
- M. Schlesinger, M. Weber, S. Schulze, M. Hietschold and M. Mehring, ChemistryOpen, 2013, 2, 146 CrossRef CAS PubMed.
- J. Wang, X. Yang, K. Zhao, P. Xu, L. Zong, R. Yu, D. Wang, J. Deng, J. Chen and X. Xing, J. Mater. Chem. A, 2013, 1, 9069 CAS.
- X. Xiao, R. Hu, C. Liu, C. Xing, C. Qian, X. Zuo, J. Nan and L. Wang, Appl. Catal., B, 2013, 140–141, 433 CrossRef CAS PubMed.
- D. K. Ma, S. M. Huang, W. X. Chen, S. W. Hu, F. F. Shi and K. L. Fan, J. Phys. Chem. C, 2009, 113, 4369 CAS.
- G. Yang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, J. Mater. Chem., 2010, 20, 5301 RSC.
- M. Gui and W. Zhang, J. Phys. Chem. Solids, 2012, 73, 1342 CrossRef CAS PubMed.
- Y. Sun, Y. Yin, B. T. Mayers, T. Herricks and Y. Xia, Chem. Mater., 2002, 14, 4736 CrossRef CAS.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429 CrossRef CAS PubMed.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112 RSC.
- X. Zhang, Z. H. Ai, F. L. Jia and L. Z. Zhang, J. Phys. Chem. C, 2008, 112, 747 CAS.
- F. Qin, G. Li, R. Wang, J. Wu, H. Sun and R. Chen, Chem.–Eur. J., 2012, 18, 16491 CrossRef CAS PubMed.
- K. Sing, D. H. Everett, R. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- J. Xu, W. Meng, Y. Zhang, L. Li and C. Guo, Appl. Catal., B, 2011, 107, 355 CrossRef CAS PubMed.
- Y. Wang, Y. Wen, H. Ding and Y. Shan, J. Mater. Sci., 2010, 45, 1385 CrossRef CAS PubMed.
- W. Wang, Y. Yu, T. An, G. Li, H. Y. Yip, J. C. Yu and P. K. Wong, Environ. Sci. Technol., 2012, 46, 4599 CrossRef CAS PubMed.
- J. Hou, C. Yang, Z. Wang, W. Zhou, S. Jiao and H. Zhu, Appl. Catal., B, 2013, 142, 504 CrossRef PubMed.
- Y. Wang, K. Deng and L. Zhang, J. Phys. Chem. C, 2011, 115, 14300 CAS.
- X. Xiao, C. Liu, R. P. Hu, X. X. Zuo, J. M. Nan, L. S. Li and L. S. Wang, J. Mater. Chem., 2012, 22, 22840 RSC.
- N. San, A. Hatipoğlu, G. Koçtürk and Z. Çınar, J. Photochem. Photobiol., A, 2002, 146, 189 CrossRef CAS.
- K. Fukui, T. Yonezawa and H. Shingu, J. Chem. Phys., 1952, 20, 722 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03200h |
| This journal is © The Royal Society of Chemistry 2015 |