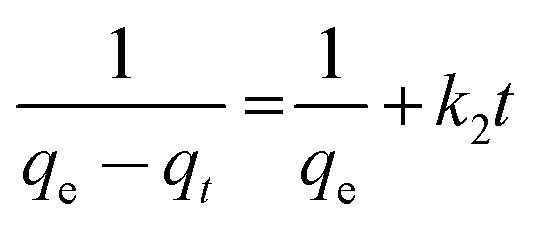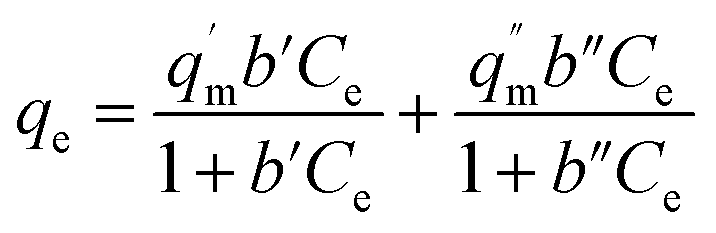DOI:
10.1039/C5RA03168K
(Paper)
RSC Adv., 2015,
5, 33023-33036
Studies on salophen anchored micro/meso porous activated carbon fibres for the removal and recovery of uranium
Received
19th February 2015
, Accepted 24th March 2015
First published on 25th March 2015
Abstract
Stringent environmental regulations emphasize the removal of uranium from aqueous systems. Activated carbon fibers (ACF) were functionalized by oxidation (ACF-OX) and salophen ligand (ACF-Sal) and evaluated for the removal of uranium. The prepared sorbents were characterized by various techniques such as scanning electron microscopy (SEM), energy dispersive X-ray (EDAX), Fourier transform infrared (FTIR) spectroscopy, Brunauer–Emmett–Teller (BET) surface area analyzer and X-ray photoelectron spectroscopy (XPS). The anchoring of salophen ligand onto ACF surface was evident from the FTIR and XPS studies. The adsorption properties of UO22+ as a function of pH and contact time were characterized by inductively coupled mass spectrometry (ICPMS). The adsorption kinetics fitted the pseudo second order kinetics and equilibrium was reached within 180 minutes. The experimental data were modelled with Langmuir and Freundlich isotherms and various isotherm parameters were evaluated. The maximum adsorption capacities of U(VI) at pH 6 for ACF, ACF-OX and ACF-Sal were found to be 22.2, 50.0 and 142.8 mg g−1, respectively. Thermodynamic studies revealed the spontaneity of the reaction and influence of other cations and anions on the sorption behaviour of uranium has been studied. Studies have been conducted to demonstrate the recyclability of the sorbent for five consecutive sorption desorption cycles. Using FTIR and XPS studies, a suitable mechanism for uranium sorption has also been postulated.
1. Introduction
Uranium is the second heaviest naturally occurring radioactive element. Uranium is the basic energy element of the present Indian nuclear power programme. It starts as a source of the fuel cycle and finally ends up as a waste component. Uranium is a well-known nephrotoxic heavy metal. It is reported that an exposure of 0.1 mg per kg body weight of natural U results in transient chemical damage to the kidneys.1 The maximum contaminant level of U(VI) in water set by the Environmental Protection Agency (EPA) is 30 μg L−1, whereas the World Health Organization's guideline value is set at 50 μg L−2. According to the Atomic Energy Research Board (AERB) of India, the maximum allowable concentration of U(VI) in water bodies is 60 μg L−1. In aquatic environments, uranium predominantly exists in its hexavalent oxidation state, e.g. UO22+. These hexavalent uranyl ions are highly mobile and migrate as stable uranyl carbonate complexes under near surface conditions.3 Cost effective remediation technology is required to tackle the removal of uranium from large volumes of wastewater. Several methods, including ion exchange, reduction, reduction followed by chemical precipitation, electrochemical precipitation, membrane separation, solvent extraction, biosorption, and adsorption, are utilized to remove uranium from wastewater and process effluents. Among the various removal technologies for U(VI) reported, adsorption is the most versatile technique owing to its ease of operation, low waste generation and considerably low recurring cost. The removal of U(VI) by various adsorbents has been adequately reviewed.4,5 Carbon based sorption materials offer various advantages such as higher radiation and thermal resistance than commonly used organic exchange resins and improved chemical stability than widely used inorganic sorbents in strongly acidic solutions in the majority of nuclear wastewaters.4 Carbonaceous materials, including activated carbon,6,7 activated carbon fibres, carbon nanotubes8–10 and mesoporous carbon,11 have been reported in the applications of U(VI) sorption. To improve the selectivity and sorption capacity towards targeted metal ions, functionalization with specific ligands are generally used.4 Among these carbonaceous materials, activated carbon fibres (ACF) are unique owing to their large porous surface area, controllable pore structure, thermo-stability and low acid/base reactivity. Furthermore, ACFs are microporous materials possessing high surface area (∼1200–1800 m2 g−1), which is a prime factor for an adsorbent. In ACF, micropores which are responsible for adsorption are connected to the external surface directly by narrow diameter fibres (10–20 μm). Thus, the diffusion length is small, resulting in negligible mass transfer coefficient and the removal rate of pollutants is adsorption controlled.12 ACFs have been found useful for the removal of variety of pollutants like Cd(II) and Pb(II),12–14 Ni(II) and Zn(II),15–17 SO2, NO and CO2,18 and 2-chloroethanol.19 Jung et al.20 have reported the electrosorption of uranium (U(VI)) ions onto a porous ACF. However, there are seldom any reports in the literature on the use of functionalized ACFs towards U(VI) removal. It is well known that salophen is a tetradentate ligand that can easily combine with uranyl cation to form a stable uranyl–salophen complex21,22 and has been found useful for the determination of trace U(VI). Thus, this study pertains to the development of functionalized activated carbon fibres by oxidation (ACF-OX), grafting of salophen ligand (ACF-Sal) and its applicability for U(VI) removal. The systematic structural characterization of the functionalized sorbents, namely, ACF-OX and ACF-Sal, were performed using various techniques and optimization of reaction parameters, including pH and reaction time, were carried out. Isotherm, thermodynamic and kinetic models were evaluated and a suitable mechanism for the adsorption of U(VI) using FTIR and XPS has also been postulated.
2. Materials and methods
All the chemicals including, sodium hydroxide (NaOH), nitric acid (HNO3), uranyl(vi) nitrate (UO2(NO3)2·6H2O), and other reagents and solvents (ethanol, acetone) used in this study were of analytical grade and all the solutions were prepared using Milli-Q purified water (resistivity > 18.2 MΩ cm). 4-Hydroxysalicylaldehyde, thionyl chloride and 1,2-diaminoethane were acquired from Sigma Aldrich chemicals. Activated carbon fibres were purchased from Nippon Kynol In. (Osaka Japan).
2.1 Preparation of ACF-OX
Around 1 g of ACF samples were treated with 20 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of conc. HNO3 and water and heated at 60 °C for 30 min, followed by a thorough washing with distilled water and dried in an oven for 12 h at 120 °C.
:1 mixture of conc. HNO3 and water and heated at 60 °C for 30 min, followed by a thorough washing with distilled water and dried in an oven for 12 h at 120 °C.
2.2 Preparation of salophen ligand
A stoichiometric amount of 4-hydroxysalicylaldehyde (0.02 mol, 2.76 g) in dissolved methanol (25 mL) was added dropwise to 1,2-diaminoethane solution (0.01 mol, 0.60 g) in 25 mL methanol. The contents were refluxed for 4 h and a bright yellow precipitate of symmetrical Schiff-base ligand, H2[(OH)2–salen], was obtained.
2.3 Preparation of ACF-Sal
Initially, chlorinated ACFs were prepared. Around 100 mg of ACFs were suspended in a solution of SOCl2 (25 mL) and DMF (1 mL). The suspension was stirred at 65 °C for 24 h. The solid was then separated by filtration, washed with anhydrous THF, and dried in vacuum. To a solution of salophen (100 mg) in degassed CHCl3 (8 mL), chlorinated ACFs were added (50 mg) and the suspension was stirred for 20 h under N2 atmosphere at 70 °C. The solid was then separated by filtration, exhaustively washed with THF and CH2Cl2, and dried under vacuum. The schematic of the preparation of ACF-OX and ACF-Sal is shown in Scheme 1.23
 |
| | Scheme 1 Schematic representation of ACF functionalization. | |
2.4 Batch studies
Adsorption experiments were carried out by a batch technique. About 0.05 g of adsorbent was placed in a beaker containing 20 mL of 0.1 to 500 mg L−1 of U(VI) solution for ACF and ACF-OX and 0.1 to 1000 mg L−1 for ACF-Sal. The pH of the solution was adjusted to 6.0 by adding 10% sodium hydroxide or 10% sulphuric acid solutions. The suspension was stirred for 3 h at an agitation speed of 110 rpm. At the end of the equilibrium time, the content was separated by filtration with a 0.22 μm pore size filter paper and U(VI) in solution was analyzed by inductively coupled plasma-mass spectrometry (ICP-MS) (Thermo Scientific, XSERIES 2). All the experiments were repeated twice. The amount of the U(VI) adsorbed (mg) per unit mass of sorbent (g), qe, was obtained by mass balance using the following equation:| | 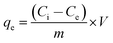 | (1) |
where Ci and Ce are initial and equilibrium concentrations of the U(VI) (mg L−1), m is dry mass of sorbent (g) and V is the volume of the solution (L). Kinetic experiments were conducted by equilibrating 20 mL of 100 mg L−1 of U(VI) at a dose rate of 5.0 g L−1 and the pH was maintained at 6.0 during equilibration. The amount of uranium adsorbed was monitored at regular time intervals. The effect of competing cations and anions were examined by maintaining the initial concentration of uranium at 100 mg L−1. Using the same conditions mentioned above, thermodynamic studies were carried out by equilibrating the solutions for 3 h at three different temperatures at 25 °C, 35 °C and 45 °C and the amount of uranium adsorbed was determined. Recyclability studies were performed using 0.1 M H3PO4 as the desorbent. After each cycle, the adsorbent was filtered and equilibrated with 20 ml of 0.1 M H3PO4 for 30 min and the adsorbent was filtered, thoroughly washed with water and used for the consequent adsorption cycle.
2.5 Analytical measurements
Fourier transform infrared (FTIR) measurements were made with KBr pellets using a Tensor 27 (Bruker, Germany) in the attenuated total reflectance (ATR) mode. A FEI Quanta 200 machine was used for scanning electron microscopy (SEM). XPS measurements were performed using a PHI 5000 Versa Prob II (FEI Inc.) spectrometer using nonmonochromatic Al Kα radiation (1486.6 eV). XPSPEAK41 software with a Gaussian–Lorentzian line shape was used for the deconvolution of individual spectral peaks. A nonlinear Shirley background subtraction was applied for fitting each spectral region. The adsorbent was analyzed for the pore size distribution (PSD), specific surface area and pore volume by N2-physisorption using a Autosorb-1C instrument (Quantachrome, USA). Uranyl ions concentrations were determined by Inductive coupled plasma mass spectroscopy ICP-MS (Thermo, X-Series2). Calibration was carried out daily with freshly prepared uranium standards before sample analysis.
3. Results and discussion
3.1 Characterization of adsorbent
3.1.1 SEM analysis.
SEM images of ACF, oxidized ACF, and ACF-Sal are shown in Fig. 1a–c, respectively. ACF are composed of bundles of fibres with a diameter of about 10 μm. It is evident from the images that the surfaces of pristine ACF, ACF-OX and ACF-Sal are found to be smooth. The loading of uranium on ACF-Sal was confirmed from the Energy Dispersive X-ray analysis (EDAX), as shown in Fig. 1d.
 |
| | Fig. 1 SEM images of (a) pristine ACF, (b) ACF-OX, (c) ACF-Sal, and (d) EDAX spectra of ACF-Sal-U. | |
3.1.2 FTIR spectra.
FTIR spectra from the ACFs show a broad peak at ∼3445 cm−1, which is a characteristic of the O–H stretch of the hydroxyl group (Fig. 2a), arising from the oscillation of carboxyl groups. The peaks at 2891 and 2920 cm−1 correspond to –CH2 and –CH symmetric stretch, respectively. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration was found at 1632 cm−1. An additional peak at 1720 cm−1 corresponding to C O stretching was found after the oxidation of ACF with nitric acid (ACF-OX) (Fig. 2b).24 In the salophen anchored ACF (Fig. 2c), the peaks at 1510 and 1620 cm−1 are attributed to the absorption of carbon–nitrogen double bond of the azomethine group.23 Furthermore, additional peaks at 1210 cm−1 and 1116 cm−1 are due to the C–O and C–N stretching vibration, respectively.21
C stretching vibration was found at 1632 cm−1. An additional peak at 1720 cm−1 corresponding to C O stretching was found after the oxidation of ACF with nitric acid (ACF-OX) (Fig. 2b).24 In the salophen anchored ACF (Fig. 2c), the peaks at 1510 and 1620 cm−1 are attributed to the absorption of carbon–nitrogen double bond of the azomethine group.23 Furthermore, additional peaks at 1210 cm−1 and 1116 cm−1 are due to the C–O and C–N stretching vibration, respectively.21
 |
| | Fig. 2 FTIR spectra of (a) plain ACF (b) ACF-OX and (c) ACF-Sal. | |
3.1.3 BET measurements.
The specific surface area of ACF, ACF-OX and ACF-Sal was obtained over the relative pressure range from 0.05 to 0.35 using the standard BET method. The total pore, mesopore and micropore volumes were calculated using the instrument's software supplied by Quantachrome using Barrett–Joyner–Halenda (BJH) and density functional theory (DFT) methods. Table 1 summarizes the data for BET surface area and pore volumes of various ACFs. The large surface area and small pore size of these materials lead to the strong confinement of the adsorbed phase, together with strong interactions with the surface. It is evident that the surface area of the ACFs increased from 1337 m2 g−1 to 1416.5 m2 g−1 upon oxidation with nitric acid and further grafting with salophen ligand does not affect the surface area of the sorbent. During oxidation, the increased surface area could be attributed to the opening of pores. The marginal decrease in pore volumes of functionalized ACFs could be attributed to the blockage of inter-bundle galleries and intra-bundle interstitial channels by various functional groups.
Table 1 Surface area and pore volumes of virgin and functionalized ACF
| Adsorbent |
Surface area (m2 g−1) |
Average diameter (nm) |
Total pore volume (cc g−1) |
Pore volume (cc g−1) |
| Meso |
Micro |
| ACF |
1337.0 |
2.10 |
0.7020 |
0.031 |
0.6320 |
| ACF-OX |
1416.5 |
0.95 |
0.6737 |
0.033 |
0.6407 |
| ACF-Sal |
1410.0 |
0.98 |
0.6800 |
0.032 |
0.6480 |
3.2 Effect of initial pH
The effect of the variation of initial pH in the range of 1–9 on adsorption using ACF, ACF-OX and ACF-Sal was examined. The initial concentration of U(VI) was maintained at 100 mg L−1. Efforts were made not to maintain the pH throughout the sorption experiments. The results obtained are shown in Fig. 3. It is evident from the figure that the sorption of U(VI) increased greatly from pH 4 to pH 6 and further increases in pH resulted in decreased sorption. At pH ≤ 3, UO22+ is the predominant species of ions and sorption is found to be very low owing to the competition of H+ ions for the active binding sites of the sorbent.9 In the pH range of 5.5–7.5, the hydrolysis of uranyl ions occurs and various multinuclear hydroxyl complexes are prevalent, including UO2(OH)+, (UO2)2(OH)22+ and (UO2)3(OH)5+.25 Thus, the maximum adsorption of U(VI) was found at pH 6. At pH values greater than 7, anionic U(VI) species (UO2)3(OH)7− were prevalent, which resulted in low sorption capacity.26 Furthermore, the amount of U(VI) increased from 18.56 mg g−1 to 35.2 mg g−1 after functionalization with salophen ligand. This can be explained by the complexation of U(VI) and salophen ligand, which are detailed in section 3.9.
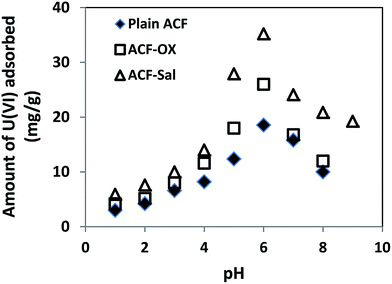 |
| | Fig. 3 Effect of initial pH on ACF, ACF-OX and ACF-Sal with U(VI). | |
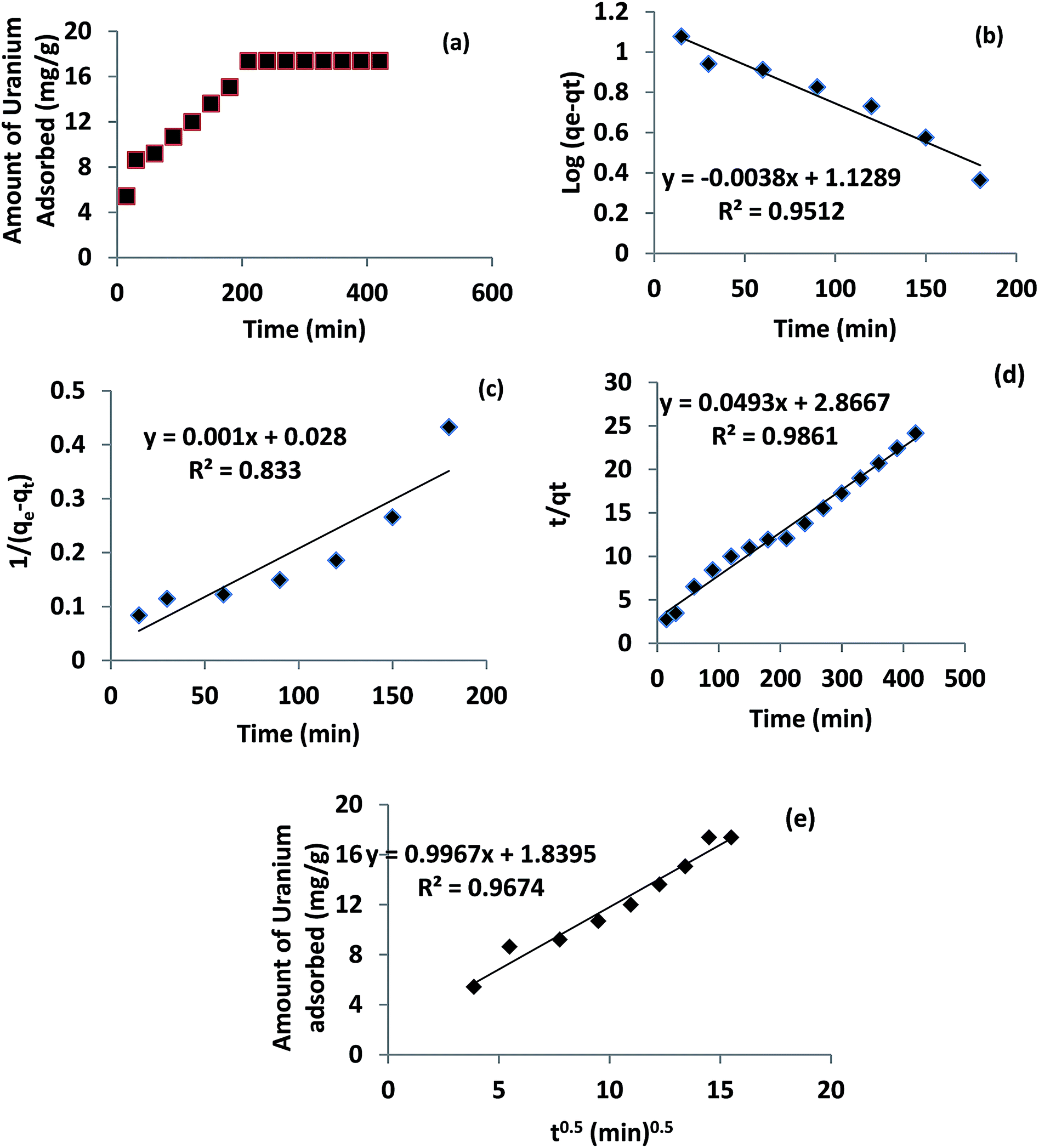
3.3 Effect of contact time
Sorption of U(VI) on ACF-Sal as a function of contact time was carried out at pH 6.0 ± 0.1. The results obtained are shown in Fig. 4a. The sorption of U(VI) on ACF-Sal was shown to be rapid and a contact time of 3.5 h was enough to reach sorption equilibrium. Thus, all the experiments were carried out at 4 h equilibration time. The kinetics of U(VI) adsorption onto ACF-Sal was modelled using the Lagergren model27 or pseudo first order, second order28 and pseudo second order,29 as shown in eqn (2)–(4), respectively.| |  | (2) |
| |  | (3) |
| |  | (4) |
where kL is the Lagergren rate constant of adsorption (min−1); k2 the second-order rate (g mg−1 min−1) and k′2 the pseudo-second-order rate constant of adsorption (g mg−1 min−1); qe and qt are the amounts of U(VI) ion (mg g−1) at equilibrium and at time t, respectively. The plots of various models are depicted in Fig. 4b–d. The rate constants obtained for various kinetic models are given in Table 2. It is evident that among the pseudo first order, second order and pseudo second order plots, the pseudo second order plot of t/qtvs. t, Fig. 4c, yielded a straight line with correlation coefficients of >0.98. Thus, it could be concluded that the sorption of U(VI) with ACF-Sal followed pseudo second order kinetics. Since this model is based on the assumption that the rate-limiting step may be chemical sorption, involving valency forces through sharing or exchange of electrons between sorbent and analyte, it is postulated that complex formation between the salophen ligand and U(VI) ions is the rate limiting step for the sorption of U(IV) onto ACF-Sal.
 |
| | Fig. 4 (a) Effect of equilibration time, (b) pseudo first order kinetic model, (c) second order kinetic model, (d) pseudo second order kinetic model, and (e) Web–Morris model of ACF-Sal and uranium system. | |
Table 2 Kinetic parameters of ACF-Sal and U(VI) systems
| Pseudo first order |
Second order |
Pseudo second order |
|
k
l (min−1) |
R
2
|
k
2 (mg g−1 min−1) |
R
2
|
k
′2 (mg g−1 min−1) |
R
2
|
| 0.0069 |
0.951 |
0.001 |
0.833 |
0.0007 |
0.986 |
It is generally known that a typical solid/liquid sorption involves film diffusion or intraparticle diffusion as well. The probability of intraparticle diffusion could be modelled by the Weber and Morris model30 and this model relates the amount of the U(VI) adsorbed and the intraparticle rate constant (kint) given by eqn (5)
| |  | (5) |
The plot of qt against √t results in an intercept (Fig. 4e). If the intraparticle diffusion is the sole rate determining step, then the plot of qtvs. √t should pass through the origin with zero intercept. However, in the present scenario (Fig. 4e), qtvs. √t does not pass through the origin. Hence, we can conclude that intraparticle diffusion is not the sole rate determining step for the sorption of U(VI) on ACF-Sal. The value of intraparticle diffusion constant (kint) of U(VI) was found to be 1.021 (g mg−1) (min0.5)−1.
3.4 Sorption isotherms
The U(VI) adsorption isotherms for ACF, ACF-OX, and ACF-Sal are presented in Fig. 5a. The adsorption data of U(VI) over various functionalized ACFs were modelled using various isotherms. The most commonly used Langmuir model describes the formation of homogeneous monolayer on the sorbent surface. The adsorption of U(VI) ions from the bulk to functionalized ACF surface could be expressed by Langmuir expression31 as follows:| |  | (6) |
where qe is the amount of U(VI) adsorbed (mg g−1) at equilibrium and Ce is the equilibrium concentration (mg L−1). The empirical constants qm and b denote the maximum adsorption capacity and energy of adsorption, respectively, and were calculated from the slope and intercept of plot between 1/Ce and 1/qe (Fig. 5b). The constant ‘b’ is attributed to the affinity between the adsorbent and analyte in the given system. The values obtained for the various constants are given in Table 3. It is evident from Fig. 5a that an “S-isotherm” curve was obtained using ACF-Sal as an adsorbent. The curve obtained is sigmoidal with a point of deflection. At low U(VI) concentrations, the adsorption is limited by the presence of the salophen ligand. After the complexation of U(VI) with the salophen ligand, normal adsorption occurs.32 The point of inflection illustrates the concentration, for which the other process of sorption overcomes the complexation. To further explain this behaviour, the data obtained was modelled by the two site Langmuir isotherm,33 given by eqn (7).| | 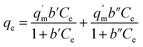 | (7) |
 |
| | Fig. 5 (a) Equilibrium isotherm, (b) linearized Langmuir plot, (c) two site Langmuir plot, (d) Freundlich plot. | |
Table 3 Isotherm parameters of ACF, ACF-OX and ACF-Sal with U(VI) systems
| Adsorbent |
Langmuir model |
Two site Langmuir model |
Freundlich model |
|
q
max (mg g−1) |
b (L mg−1) |
R
2
|
q
′m (mg g−1) |
b′ (L mg−1) |
R
2
|
q
′′m (mg g−1) |
b′′ (L mg−1) |
R
2
|
K
F (L g−1) |
| ACF |
22.22 |
0.8640 |
0.96 |
— |
— |
— |
— |
— |
— |
2.673 |
| ACF-OX |
50.00 |
0.0149 |
0.94 |
— |
— |
— |
— |
— |
— |
2.138 |
| ACF-Sal |
142.86 |
0.0029 |
0.99 |
204.08 |
0.0023 |
1.00 |
101.01 |
0.0043 |
0.99 |
0.916 |
The two-site Langmuir isotherms fit the adsorption data well when there are two types of adsorption sites with different binding energies on the adsorbents.33q′m (mg g−1) and q′′m (mg g−1) are the maximum absorption capacities and b1 (L mg−1) and b2 (L mg−1) in eqn (7) are the affinity coefficients to sites 1 and 2 on the adsorbents, respectively. The total maximum adsorption capacity can be obtained by adding q′m and q′′m. It is evident from the data that the maximum adsorption capacity of the three sorbents towards U(VI) is in the order ACF-Sal > ACF-OX > ACF. It is evident that the sorption capacity of ACF-Sal from the two site Langmuir model is 13.68 times higher than plain ACF. The high sorption capacity of ACF-Sal towards U(VI) could be attributed to the complex formation between salophen ligand and U(VI) ions. The adsorption capacity of ACF-Sal is significantly higher than the adsorption capacity of various carbon adsorbents such as plain oxidized MWCNT26 (43.32 mg g−1), carboxymethyl cellulose grafted CNT34 (112.0 mg g−1), diglycolamide functionalized MWCNTs35 (133.7 mg g−1), palm shell activated carbon36 (51.81 mg g−1) and imine functionalized carbon spheres37 (113 mg g−1).
Further analysis of the Langmuir model could be achieved based on a dimensionless equilibrium parameter called separation factor (RL):38
| |  | (8) |
where
C0 is the initial concentration of U(
VI) and ‘
b’ is the Langmuir adsorption equilibrium constant (mL mg
−1). The value of
RL indicates that the isotherm represents a favourable (0 <
RL < 1), unfavourable (
RL < 1), linear (
RL = 1), or irreversible process (
RL = 0). For an initial concentration of 100 mg L
−1, the U(
VI)
RL values for ACF, ACF-OX and ACF-Sal were found to be 0.0115, 0.4016, and 0.7752, respectively. These values suggest that the adsorption of uranyl ions by ACF and functionalized ACFs is a favourable process.
The heterogeneity of the system is described by the Freundlich model and its linearized form is represented below39
| | 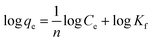 | (9) |
Fig. 5d denotes the Freundlich plots of various sorbents. n and Kf are the Freundlich parameters, which represent the adsorption capacity and adsorption intensity, respectively. For a good adsorbent, the values of n ranged between 1 and 10. It is evident from Table 3 that the ‘n’ values ranged between 1.335 and 2.024 with a regression coefficient of 0.93 to 0.98, indicating the strong interaction of U(VI) ions with the adsorbent.
3.5 Thermodynamic studies
The commonly used thermodynamic parameters, such as ΔG0, ΔH0 and ΔS0, were calculated from the adsorption data.40 Initially, Kc, the equilibrium constant, was determined by eqn (10)| | 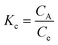 | (10) |
where CA (g L−1) is the concentration of solute in the aqueous phase and Ce is the equilibrium concentration (g L−1). ΔG0 was calculated using the following equation:| | | ΔG0 = −RTlnKc | (11) |
where R is the gas constant and T is the temperature in Kelvin. Using the Van't Hoff eqn (12) the value of ΔS and ΔH was determined:| | 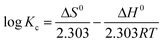 | (12) |
Based on the above-calculated data, a linear plot of lnKcvs. 1/T was drawn for the U(VI) and ACF-Sal system (Fig. 6). Using these plots, ΔS0 and ΔH0 were determined from the intercept and slope, respectively. The data obtained are presented in Table 4. The negative free energy values indicate the spontaneity and feasibility of the process, while the positive ΔH0 values indicate the endothermic nature of the process. Furthermore, positive values of the entropy (ΔS0) of adsorption could be attributed to metal ion dehydration due to surface sorption on ACF-Sal.
 |
| | Fig. 6 Thermodynamic studies of ACF-Sal and U(VI) systems. | |
Table 4 Thermodynamic parameters
|
T (K) |
C
e (g L−1) |
K
c
|
ΔG (kJ mol−1) |
ΔS (J mol−1) |
ΔH [kJ (mol−1 K−1)] |
| 298 |
0.2476 |
1.0192 |
−0.0472 |
|
|
| 308 |
0.2309 |
1.1649 |
−0.3909 |
4.439 |
10.96 |
| 318 |
0.2131 |
1.3463 |
−0.7863 |
|
|
3.6 Recyclability studies
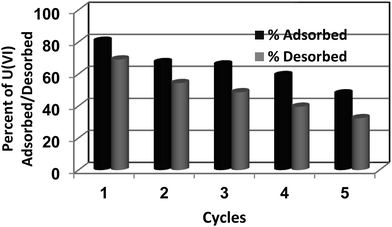
Uranium desorption studies were conducted after the sorption of U(VI) using ACF-Sal as a sorbent. The conditions for adsorption were maintained as prescribed in Sec 2.4. After the adsorption of U(VI) ions, the sorbents were filtered, washed with water, and 0.1 M H3PO4 was used as a desorbent. The sorption–desorption cycle was repeated for 5 cycles. The amount of U(VI) adsorbed by ACF-Sal for five consecutive sorption–desorption cycles are depicted in Fig. 7. It is evident from the figure that around a 20% and 40% decrease in U(VI) uptake was observed at the end of the 3rd and 5th cycle, respectively. A similar result has been reported using ordered mesoporous carbon as an adsorbent.11
 |
| | Fig. 7 Recyclability studies. | |
3.7 Effect of competing ions
Uranium sorption by ACF-Sal in the presence of other cations and anions was studied. Initially, 100 ppm of U(VI) was spiked with known concentrations of anions/cations and pH was adjusted to 6 and equilibrated for 3 h. After equilibration, the solution was filtered and the amount of U(VI) adsorbed was analyzed using an ICP-MS instrument. The effect of these ions on the sorption process may be represented by the ratio of adsorption capacity in the presence of interfering ion (qmix) and without interfering ion (q0), as shown below.41
q
mix/q0 > 1, increased adsorption in the presence of other interfering ions; qmix/q0 = 1, adsorption is not influenced in the presence of other interfering ions; qmix/q0 < 1, adsorption is suppressed in the presence of other interfering ions.
The effect of cations and anions on the sorption capacity of ACF-Sal is detailed in Tables 5 and 6, respectively. The order of interfering effect of the various cations tested is Cd2+ > Ni2+ > Pb2+ > Cu2+ > Ca2+ ⋙ Mn2+. An increased interfering effect was observed for Cd2+, Ni2+ and Pb2+ ions. This could be attributed to their complexation with salophen ligand and non-availability of these complexing sites for U(VI) sorption. Among the various anions tested, it is interesting to observe that a slight increase of the sorption of U(VI) ions is observed in the presence of phosphate ions. Coordination around uranium is pentagonal bipyramidal with the four donor atoms of the salophen ligand occupying the equatorial plane and the uranyl oxygen atoms in the axial positions. The fifth equatorial site of the metal remains available for coordination by an additional group, either an anion or a neutral molecule. Due to the high affinity of UO22+ to phosphate anions,42 the stability constants of Sal–UO2–H2PO4− is larger than other anions (Cl−, SO42−, NO3−) tested and the data is furnished in Table 7. This explains the increased uptake of U(VI) by ACF-Sal in the presence of phosphate ions. Furthermore, it should be noted that when desorption studies were conducted with 0.1 M H3PO4, around 85% of U(VI) was found to be desorbed (Sec 3.6). This is probably because the acidity of the desorbing solution is around pH 1 and at this acidity, the U(VI) salophen complex is unstable and uranyl–phosphate complexation takes place, which results in leaching of U(VI) from the sorbent surface.
Table 5 Effect of competing cations on sorption of U(VI) by ACF-Sal
| Cations |
q
mix/q0 |
| 100 mg L−1 |
200 mg L−1 |
| Ni2+ |
0.91 |
0.89 |
| Cd2+ |
0.82 |
0.76 |
| Ca2+ |
0.97 |
0.94 |
| Pb2+ |
0.90 |
0.86 |
| Cu2+ |
0.99 |
0.92 |
| Mn2+ |
1.00 |
1.00 |
Table 6 Effect of competing anions on sorption of U(VI) by ACF-Sal
| Anions (0.1 M) |
q
mix/q0 |
| Sulphate |
0.97 |
| Chloride |
0.93 |
| Nitrate |
1.00 |
| Phosphate |
1.10 |
Table 7 The experimental stability constants (Kass) for the uranyl salophene (US1) complexation with anionsa
| Complex |
K
ass [M−1] |
|
Adapted from ref Stauthammer, W. Ph.D. Thesis, University of Twente, The Netherlands, 1994.
|
| Sal–UO2–Cl− |
4.5 × 102 |
| Sal–UO2–NO2− |
3.1 × 102 |
| Sal–UO2–HSO4− |
5.0 × 101 |
| Sal–UO2–H2PO4− |
1.1 × 104 |
3.8 XPS evaluation
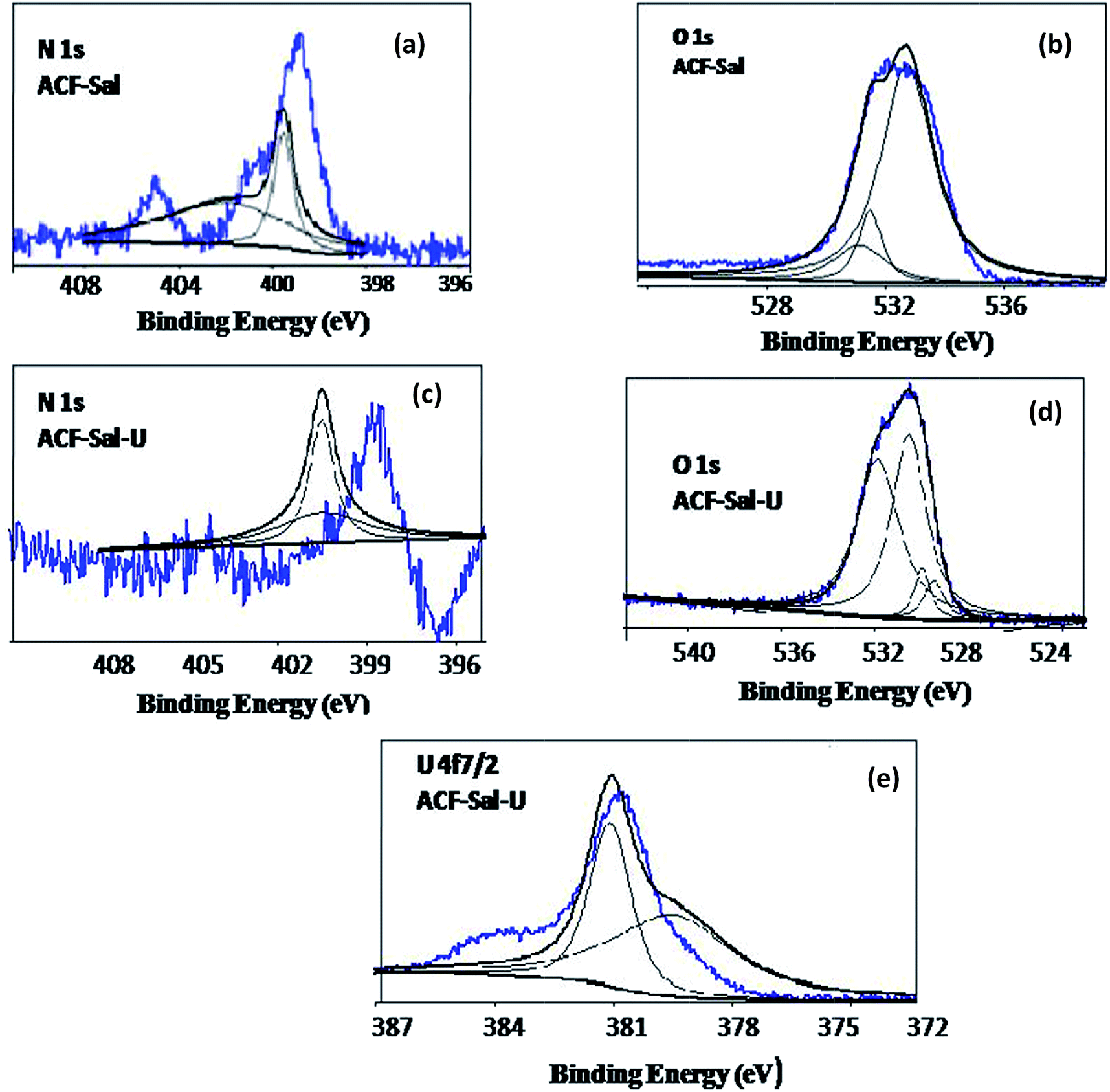
In order to study the sorption of U(VI) onto ACF, ACF-OX and ACF-Sal, XPS spectra were recorded after U(VI) loading. The O 1s, C 1s, and U 4f of the sorbents are demonstrated in Fig. 8. In the survey spectra (Fig. 8), the characteristic doublet peaks of U 4f5/2 and U 4f7/2 were observed for all the loaded sorbents. The binding energies of the various peaks and the splitting values of uranium peaks are depicted in Table 8. The peak positions of U(VI) adsorbed on ACF-Sal shifted to relative high binding energies as compared to those of U(VI) adsorbed onto ACF and ACF-OX, which could be attributed to the stronger interaction of U(VI) with ACF-Sal compared to ACF and ACF-OX. A similar observation was observed by other researchers on uranyl sorption onto chitosan modified CNTs.43 Upon further comparison of the splitting values of 4f5/2 and 4f7/2 peaks of U(VI) ions onto ACF, ACF-OX, and ACF-Sal, an increasing trend is observed, which could be attributed to stronger interaction of U(VI) with salophen ligand.
 |
| | Fig. 8 Uranium loaded XPS wide scans of ACF, ACF-OX and ACF-Sal. | |
Table 8 Binding energies (eV) of ACF, ACF-OX and ACF-Sal after uranium adsorption
| Type |
C 1s |
O 1s |
N 1s |
U 4f5/2 |
U 4f7/2 |
Splitting values |
| ACF-U |
284.35 |
530.80 |
— |
390.87 |
381.00 |
9.87 |
| ACF-OX-U |
284.48 |
530.43 |
— |
391.57 |
381.25 |
10.32 |
| ACF-Sal-U |
285.30 |
531.35 |
398.53 |
391.67 |
381.30 |
10.37 |
In order to further probe the mechanism of U(VI) sorption on to ACF-Sal at the molecular level, the XPS spectra of survey and high resolution scans for O 1s, N 1s, and U 4f on ACF-Sal were recorded. The O 1s, N 1s, and U 4f of the ACF-Sal before and after U(VI) sorption (denoted as ACF-Sal-U) are demonstrated in Fig. 9. The peak fitting results of the U 4f, O 1s, and N 1s before and after U(VI) sorption on ACF-Sal are listed in Table 9. As shown in Fig. 9e, the U 4f7/2 spectrum was resolved into two peaks: the peak at 379.5 eV corresponded to the free uranyl adsorbed on ACF-Sal, and the peak at 381.08 eV was attributed to covalent bond of azomethine N–U(VI).44Fig. 9b shows that the O 1s spectra could be resolved into three main peaks, occurring at 530.54, 531.94 and 532.77 eV corresponding to OC, C–O–C and H–O–H bonds,45 respectively. Thus, XPS studies have been used to study the efficient anchoring of salophen ligand onto the CNT back bone.46 After U(VI) sorption (Fig. 9d) four different peaks occurred at 529.47, 529.97, 530.53, 531.85 eV. An additional peak after U(VI) sorption could be attributed to the presence of the UO bond.46,47 Furthermore, the shift in the binding energies of O 1s before and after U(VI) loading indicated that U(VI) sorption onto ACF-Sal occurred by the complexation of oxygen-containing functional groups. The calculation of the content of elements on the surface of ACFs by the area of each element showed that the weight content of N is about 3.2%, indicating the presence of Schiff-base groups. Fig. 9a and c show the core level N 1s of ACF-Sal before and after U(VI) sorption, respectively. The N 1s spectrum was resolved into two individual component peaks at 395.5 and 402.0 eV.48 After uranyl sorption, the peaks shifted to higher binding energies due to the charge transfers occurring from nitrogen containing salophen ligand to U(VI) ions.
 |
| | Fig. 9 Curve fitted high resolution XPS scans of ACF-Sal for (a) O 1s, (b) N 1s before U(VI) loading and (c) O 1s, (d) N 1s and U 4f (e) after U(VI) loading. | |
Table 9 Core level binding energies of ACF-Sal and U(VI) loaded ACF-Sal systems
| Core levels |
ACF-Sal |
ACF-Sal-U |
| N 1s |
Binding energy (eV) |
FWHW (eV) |
Area |
Binding energy (eV) |
FWHW (eV) |
Area |
| 395.5 |
1.00 |
143.38 |
400.50 |
3.520 |
82.12 |
| 402.0 |
6.386 |
336.99 |
406.09 |
0.592 |
0.59 |
| O 1s |
530.54 |
1.69 |
2326.78 |
531.85 |
2.230 |
2154.16 |
| 531.94 |
1.37 |
982.99 |
530.53 |
1.766 |
2002.55 |
| 532.77 |
1.51 |
186.71 |
529.47 |
1.000 |
243.86 |
| — |
— |
— |
529.97 |
0.810 |
252.97 |
| U 4f7/2 |
— |
— |
— |
379.5 |
3.543 |
1148.07 |
| — |
— |
— |
381.08 |
1.263 |
735.34 |
Thus, the results from XPS studies suggest that the salophen ligand is efficiently anchored onto the ACF surface and U(VI) complexation occurred with the tetradentate N(2)O(2) donors derived from the phenolic oxygen and azomethine nitrogen of the saplohen ligand.
3.9 Mechanism of interaction
Adsorption of U(VI) onto ACF and ACF-OX might be attributed to both ion exchange and electron donating acceptor complexation reactions at the surface sites. At pH 6.0, where in the maximum removal of U(VI) is observed (Fig. 3), 95% of the uranyl ions exist as (UO2)3(OH)5+ species and few as UO22+.49 It is well known that at pH 6.0, the carboxyl groups are deprotonated and there exists a strong complexation between the hydrolyzed uranyl ions [(UO2)3(OH)5+] and carboxyl groups. The FTIR spectra of ACF, ACF-OX and ACF-Sal before and after loading with U(VI) are shown in Fig. 10. It is evident from the spectra that after U(VI) loading in ACF (Fig. 10a), the hydroxyl peak shifts from 3437 cm−1 to 3418 cm−1, which confirms the involvement of OH group and a sharp peak at 920 cm−1 confirms the υ3 band of uranyl ions.50,51 In U(VI) loaded ACF-OX, the shifting of the carbonyl stretching peak from 1721 to 1715 cm−1 is observed. In addition, a uranyl band appears at 906 cm−1, which is a characteristic of the υ3 band of uranyl ions described earlier. Moreover, the U(VI) removal might be attributed to the electron donating acceptor (EDA) complexation between the de-localized π-electron of graphene layers of ACF and (UO2)3(OH)5+ by dispersive forces.52
 |
| | Fig. 10 FTIR spectra of (a) ACF (b) ACF-OX and (c) ACF-Sal before and after loading uranyl ions. | |
The main mechanism governing the sorption of U(VI) onto ACF-Sal is the complexation reaction occurring between the salophen ligand and the uranyl ions. After complexation with U(VI) (Fig. 10c), the environment of CN changes and a shift from 1620 to 1615 cm−1 is observed. Furthermore, the stretching vibrations of –C–O and C–N exhibited a slight upfield shift to 1220 cm−1 and 1124 cm−1, respectively, owing to the complexation.21 As observed in ACF and ACF-OX, a υ3 uranyl band appeared around 890 cm−1 for U(VI) loaded ACF-Sal. The shifting of the frequencies of the uranyl band depends on the ligands present in the equatorial plane. In general, the asymmetrical uranyl stretching frequency ranges from 885 cm−1 to 899 cm−1 for uranyl complexes with Schiff base.53–57 This further confirms the complexation between U(VI) and salophen ligand.
Based on the discussions of the FTIR and XPS studies, a mechanism has been suggested in Scheme 2, depicting the complexation of uranium and salophen ligand.
 |
| | Scheme 2 Schematic of complexation of U(VI) with ACF-salophen. | |
4. Conclusions
Functionalized activated carbon fibres were prepared by oxidation and grafting salophen ligand. The prepared sorbent materials were characterized by various spectral techniques. The kinetics followed a pseudo second order model and maximum removal of U(VI) was found at pH 6. The experimental data obtained were analyzed by various isotherms, including Langmuir and Freundlich. The adsorption capacities of the functionalized ACFs were in the order of ACF-Sal > ACF-OX > ACF. The observed high capacity (142.8 mg g−1 for ACF-Sal by Langmuir and 305.09 mg g−1 by two site Langmuir model) could be attributed to the bonding of U(VI) and the salophen ligand. The effect of other commonly occurring cations and anions on the sorption U(VI) by ACF-Sal was studied. Thermodynamic studies revealed the spontaneity of the reaction and the sorbent could be recycled for 5 sorption–desorption cycles. From the FTIR and XPS studies a suitable mechanism for sorption has also been proposed.
Acknowledgements
Authors thank the funding received from the Board of Research in Nuclear Sciences, Department of Atomic Energy, Mumbai, India (Ref. no. 2013/36/57-BRNS/2482) to carry out this study.
Notes and references
-
G. M. Naja and B. Volesky, Toxicity and sources of Pb, Cd, Hg, Cr, As and radionuclides in the environment, CRC Press, Taylor & Francis Group, USA, 2009, p. 16 Search PubMed.
-
WHO, Guidelines for Drinking Water Quality, Geneva, 2nd edn, 1998, p. 283 Search PubMed.
- A. Kilincarslan and S. Akyil, J. Radioanal. Nucl. Chem., 2005, 264, 541 CrossRef CAS.
- P. D. Bhalara, D. Punetha and K. Balasubramanian, J. Environ. Chem. Eng., 2014, 2, 1621 CrossRef CAS.
- P. Swain, C. Mallika, R. Srinivasan, U. K. Mudali and R. Natarajan, J. Radioanal. Nucl. Chem., 2013, 298, 781 CrossRef CAS.
- A. M. A. Morsy and A. E. M. Hussein, J. Radioanal. Nucl. Chem., 2011, 288, 341 CrossRef CAS.
- M. Caccin, F. Giacobbo, M. Da Ros, L. Besozzi and M. Mariani, J. Radioanal. Nucl. Chem., 2013, 297, 9 CrossRef CAS.
- Y. Sun, S. Yang, G. Sheng, Z. Gua and X. Wang, J. Environ. Radioact., 2012, 105, 40 CrossRef CAS PubMed.
- A. Schierz and H. Zanker, Environ. Pollut., 2009, 157, 1088 CrossRef CAS PubMed.
- D. D. Shao, Z. Q. Jiang, X. K. Wang, J. X. Li and Y. D. Meng, J. Phys. Chem., 2009, 113, 860 CrossRef CAS PubMed.
- B.-W. Nie, Z.-B. Zhang, X.-H. Cao, Y.-H. Liu and P. Liang, J. Radioanal. Nucl. Chem., 2012, 295, 663 CrossRef.
- V. Gaur, A. Sharma and N. Verma, Chem. Eng. Process., 2006, 45, 1–13 CrossRef CAS.
- B. M. Babić, S. K. Milonjić, M. J. Polovina, S. Čupić and B. V. Kaludjerović, Carbon, 2002, 40, 1109 CrossRef.
- R. Leyva-Ramos, M. S. Berber-Mendoza, J. Salazar-Rabago, R. M. Guerrero-Coronado and J. Mendoza-Barron, Adsorption, 2011, 17, 515 CrossRef CAS.
- J. R. Rangel-Mendez and M. Streat, Water Res., 2002, 36, 1244 CrossRef CAS PubMed.
- M. A. Alvarez-Merino, V. Lopez-Ramon and C. Moreno-Castilla, J. Colloid Interface Sci., 2005, 288, 335 CrossRef CAS PubMed.
- M. S. Berber-Mendoza, R. Leyva-Ramos, F. J. Cerino-Cordoba, J. Mendoza-Barron, H. J. Amezquita Garcia and J. V. Flores-Cano, Water, Air, Soil Pollut., 2013, 224, 1604 CrossRef.
- X. Zhou, H. Yi, X. Tang, H. Deng and H. Liu, Chem. Eng. J., 2012, 200–202, 399 CrossRef CAS.
- M. Bikshapathi, S. Mandal, G. N. Mathur, A. Sharma and N. Verma, Ind. Eng. Chem. Res., 2011, 50, 13092–13104 CrossRef CAS.
- C.-H. Jung, H.-Y. Lee, J.-K. Moon, H.-J. Won and Y.-G. Shul, J. Radioanal. Nucl. Chem., 2011, 287, 833 CrossRef CAS.
- M. Wu, L. Liao, M. Zhao, Y. Lin, X. Xiao and C. Nie, Anal. Chim. Acta, 2012, 729, 80 CrossRef CAS PubMed.
- J. L. Sessler, P. J. Melfi and G. D. Pantos, Coord. Chem. Rev., 2006, 250, 816 CrossRef CAS.
- M. Salavati-Niasari and M. Bazarganipour, Appl. Surf. Sci., 2008, 255, 2963–2970 CrossRef CAS.
- C.-C. Huang and Y.-J. Su, J. Hazard. Mater., 2010, 175, 477 CrossRef CAS PubMed.
- G. Wang, J. Liu, X. Wang, Z. Xie and N. Deng, J. Hazard. Mater., 2009, 168, 1053–1058 CrossRef CAS PubMed.
- M. Wang, J. Qiu, X. Tao, C. Wu, W. Cui, Q. Liu and S. Lu, J. Radioanal. Nucl. Chem., 2011, 288, 895 CrossRef CAS.
- S. Lagergren, K. Sven. Vetenskapsakad. Handl., 1898, 24, 1–39 Search PubMed.
- Y. S. Ho, D. A. J. Wase and C. F. Forster, Environ. Technol., 1996, 17, 71 CrossRef CAS.
-
Y. S. Ho and G. McKay, Advances in Adsorption Separation Science and Technology, South China University of Technology Press, Guangzhou, 1997, pp. 257–263 Search PubMed.
- W. J. Weber and J. C. Morris, J. Sanit. Eng. Div., Am. Soc. Civ. Eng., 1963, 89, 31 Search PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361 CrossRef CAS.
- G. Sposito, On the use of the Langmuir equation in the interpretation of “adsorption” phenomena: II. The “two surface” Langmuir equation, Soil Sci. Soc. Am. J., 1982, 46, 1147–1152 CrossRef CAS.
- Y. Jin, F. Liu, M. Tong and Y. Hou, J. Hazard. Mater., 2012, 227, 461 CrossRef PubMed.
- D. Shao, Z. Jiang, X. Wang, J. Li and Y. Meng, J. Phys. Chem. B, 2009, 113, 860 CrossRef CAS PubMed.
- A. K. S. Deb, P. Ilaiyaraja, D. Ponraju and B. Venkatraman, J. Radioanal. Nucl. Chem., 2012, 291, 877 CrossRef CAS.
- Z.-J. Yi, J. Yao, J.-S. Xu, M.-S. Chen, W. Li, H.-L. Chen and F. Wang, J. Radioanal. Nucl. Chem., 2014, 301, 695 CrossRef CAS.
- P. S. Dubey, D. A. Dwivedi, M. Sillanpaa, Y.-N. Kwon and C. Lee, RSC Adv., 2014, 4, 46114 RSC.
- N. Sankararamakrishnan, A. Dixit, L. Iyengar and R. Sanghi, Bioresour. Technol., 2006, 97, 2377 CrossRef CAS PubMed.
- H. M. F. Freundlich, J. Phys. Chem., 1906, 57, 385 CAS.
-
P. Perrot, A to Z of Thermodynamics, Oxford University Press, New York, 1998 Search PubMed.
- F. V. Pereira, L. V. A. Gurgel and L. F. Gil, J. Hazard. Mater., 2010, 176, 856 CrossRef CAS PubMed.
- M. Merdivan, M. B. Buchmeister and G. Bonn, Anal. Chim. Acta, 1999, 402, 91 CrossRef CAS.
- J.-H. Chen, D.-Q. Lu, B. Chen and P.-K. Ouyang, J. Radioanal. Nucl. Chem., 2013, 295, 2233 CrossRef CAS.
- W. Song, M. Liu, R. Hu, X. Tan and J. Li, J. Chem. Eng., 2014, 246, 268 CrossRef CAS.
- M. Salavati-Niasari, F. Davar and M. Bazarganipour, Dalton Trans., 2010, 39, 7330 RSC.
- S. Vanden Berghe, F. Miserque, T. Gouder, B. Gaudreau and M. Verwerft, J. Nucl. Mater., 2001, 294, 168 CrossRef CAS.
- S. Chen, J. Hong, H. Yang and J. Yang, J. Environ. Radioact., 2013, 126, 253 CrossRef CAS PubMed.
- R. J. J. Jansen and H. van Bekkum, Carbon, 1995, 33, 1021 CrossRef CAS.
- E. Guibal, C. Roulph and P. Le Cloirec, Water Res., 1992, 26, 1139 CrossRef CAS.
- R. L. Frost, Spectrochim. Acta, Part A, 2006, 64, 308 CrossRef PubMed.
- M. Tsezos and B. Volesky, Biotechnol.
Bioeng., 1982, 24, 385 CrossRef CAS PubMed.
- S. M. Yakout, S. S. Metwally and T. El-Zakla, Appl. Surf. Sci., 2013, 280, 745 CrossRef CAS.
- K. Mizuoka, S.-Y. Kim, M. Hasegawa, T. Hoshi, G. Uchiyama and Y. Ikeda, Inorg. Chem., 2003, 42, 1031 CrossRef CAS PubMed.
- K. Mizuoka, S. Tsushima, M. Hasegawa, T. Hoshi and Y. Ikeda, Inorg. Chem., 2005, 44, 6211 CrossRef CAS PubMed.
- K. Mizuoka and Y. Ikeda, Radiochim. Acta, 2004, 92, 631 CrossRef CAS.
- L. Cattalini, S. Degetto, M. Vidali and P. A. Vigato, Inorg. Chim. Acta, 1972, 6, 173 CrossRef CAS.
- E. M. Nour, A. A. Taha and I. S. Alnaimi, Inorg. Chim. Acta, 1988, 141, 139 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence