
A biocompatible process to prepare hyaluronan-based material able to self-assemble into stable nano-particles†
Abstract
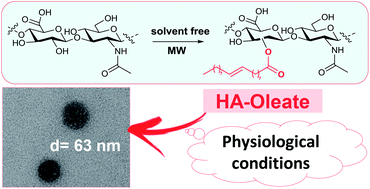
New self-assembled nano-particles were developed by chemical conjugation of natural fatty acids to the backbone of hyaluronan (HA). The chemical structure and the self-association behavior of them were studied by FT-IR, NMR, fluorescence and dynamic light scattering. The HA derivatives form stable spherical shape aggregates, as assessed by transmission electron microscopy.
 Please wait while we load your content...
Please wait while we load your content...

