
Highly dispersed MnOx nanoparticles supported on three-dimensionally ordered macroporous carbon: a novel nanocomposite for catalytic reduction of NOx with NH3 at low temperature†
Xin Gaoab,
Ling Lib,
Lihong Songab,
Ting Lub,
Jiaxin Zhaob and
Zhi Liu*ab
aInstitute of Chemistry for Functionalized Materials, Faculty of Chemistry and Chemical Engineering, Liaoning Normal University, Dalian 116029, China. E-mail: zhiliu@lnnu.edu.cn; Fax: +86 411 82156858; Tel: +86 411 82156989
bFaculty of Chemistry and Chemical Engineering, Liaoning Normal University, Dalian, Liaoning 116029, China
First published on 18th March 2015
Abstract
In this paper, we report a novel nanocomposite of MnOx nanoparticles supported by three-dimensionally ordered macroporous carbon (MnOx/3DOMC) fabricated by means of a simple multi-component infiltration of three-dimensional templates and its use as a catalyst for low-temperature selective catalytic reduction (SCR) of NOx with NH3. Several techniques, including scanning electron microscopy, X-ray diffraction, N2-sorption, transmission electron microscopy, X-ray photoelectron spectroscopy, NH3 temperature-programmed desorption, and H2 temperature-programmed reduction, are employed to characterize the MnOx/3DOMC nanocomposite. The results demonstrate that MnOx/3DOMC possesses a highly ordered macroporous structure with hierarchical mesopores in the walls of the macroporous skeleton. MnOx nanoparticles of 2–4 nm are observed to be highly dispersed on the 3DOM carbon scaffold. Compared with the MnOx/NAC and MnOx/TiO2 catalysts prepared by a conventional impregnation method, the MnOx/3DOMC catalyst exhibits better low-temperature NH3-SCR activity, stability, and water vapor and/or SO2 resistance ability. Such material represents a promising exploratory direction for enhancing the catalytic performance of metal oxide-based NH3-SCR catalysts.
Introduction
Nitrogen oxides (NOx, x = 1 or 2), which are mainly emitted from fossil fuel combustion and automobiles, are major hazardous gases for air pollution and human health.1,2 Ammonia selective catalytic reduction (NH3-SCR) of NOx to N2 and H2O has been successfully developed and commercialized as the most effective flue gas cleaning technology for stationary sources due to its high-efficiency of NOx conversion. So far, vanadium-based catalysts, which have exhibited high catalytic activity, selectivity, and SO2 tolerance within the narrow temperature window of 300–400 °C, are widely used commercial catalysts for the NH3-SCR of NOx.3,4 However, the temperature of the flue gas after the desulfurizer and particulate removal device is generally below 200 °C. Therefore, it is highly desirable to design and fabricate new low-temperature catalysts for the NH3-SCR of NOx. Over the past few years, manganese oxides (MnOx)-based catalysts have been found to be active for the NH3-SCR of NOx.5,6 Recent literature results showed that various MnOx-based catalysts, such as, nano-MnOx,7,8 MnOx/mixed-oxides,9,10 MnOx/ZSM-5,11 MnOx/TiO2,12–14 MnOx/CeO2,15 MnOx/activated carbon,16 MnOx/carbon nanotubes,17,18 etc., also presented a high NOx conversion at low temperature (≦250 °C) in the NH3-SCR reaction. However, enlarging specific surface area of the support, dispersing the catalytically active species as highly as possible, and realizing an optimized utilization of active sites are still challenging for these MnOx-based catalysts.Three-dimensionally ordered macroporous (3DOM) materials, with uniform pore size and well-defined periodic structure, have triggered increasing interests due to their widespread applications in separation, catalysis, energy storage/conversion systems, and solar cells, etc.19–25 Conventionally, the colloidal crystal templating strategy (CCTS) is employed to prepare 3DOM materials.26 Briefly, uniform monodispersed microspheres, such as polymethyl methacrylate (PMMA), polystyrene (PS) or silica spheres, can assemble into ordered three-dimensional array in densified packing. These ordered arrays offer a 3D scaffold in which a variety of precursors can be infiltrated. After subsequent solidification of the precursors and removal of the colloidal microspheres, periodic 3D framework structures can be obtained successfully. However, the single macroporous size distributions and relatively low surface areas are greatly disadvantageous to the efficient application of the 3DOM materials especially when they are used as supports in catalysis field. In order to address the problems, a upgraded dual ‘hard-soft’ colloidal crystal templating strategy (dual-CCTS) has been recently proposed to prepare hierarchically porous structure 3DOM materials, in which PMMA or PS microspheres were used as the hard template to create macropores and some surfactants or organics were introduced into the precursors as the soft template to form mesopores.27–33 It is widely accepted that the metal-support interaction and active species dispersion are important factors in the determination of catalytic performances for metal-loaded catalysts. And a larger specific surface area of the support permits higher dispersion of the active phase, which consequently leads to promotional catalytic performance.34–36 Obviously supports such as hierarchical 3DOM materials not only possess high surface area for good active species dispersion, but it can also reduce the diffusion resistance and permit facile transport, thus allowing for the efficient mass transport of the reactant and the product molecules. Moreover, desired active species can be directly introduced into the 3DOM solid-state architecture through using the corresponding metal salt solution(s) as precursor(s) to infiltrate the template. Different from the traditional impregnation, deposit or (co-)precipitation, such a 3DOM catalyst would not only yield intimate contact and strong interaction between the active species and the 3DOM support matrix, but it also make the active species highly dispersed in the 3DOM support matrix, which will potentially give rise to superior catalytic performance.
In the present work, we have successfully prepared a MnOx/3DOM carbon nanocomposite (MnOx/3DOMC) by means of a simple multi-component (manganous nitrate, phenol-formaldehyde resol, and triblock copolymer F127) infiltration of 3D PMMA templates followed by a direct pyrolysis process. The NH3-SCR of NOx was used as a probe reaction to evaluate the catalytic performance of the MnOx/3DOMC. The results showed that the MnOx nanoparticles prepared with the dual-CCTS were highly dispersed in the 3DOM carbon scaffold, and the new nanocomposite presented a higher catalytic activity than those of the others MnOx-based catalysts prepared by the conventional impregnation method in the NH3-SCR of NOx.
Experimental
Synthesis of PMMA template
The PMMA colloidal crystal templates were synthesized via a soap-free-emulsion polymerization according to a modified procedure based on ref. 37. In a typical synthesis, a required amount of monomer MMA was washed three times with a NaOH solution (10 wt%) and di-distilled water respectively to remove any traces of the inhibitor. Then, 11.3 g of the washed MMA and 150 g of di-distilled water were added to a 500 mL round bottom flask and stirred for a while. Then, 12 mL (0.0005 g mL−1) K2S2O8 was added to the solution and the mixture was constantly reacted at 70 °C for 7 h under a nitrogen atmosphere with mild stirring until a milky solution appeared. After filtering the milky solution to remove large agglomerates, the monodispersed PMMA microspheres were obtained. Finally, the as-synthesized monodispersed PMMA microspheres were packed into PMMA colloidal crystal template by evaporating the water solvent at 70 °C for 24 h.Preparation of precursors
The phenol-formaldehyde (P-F) resol precursor was prepared according to ref. 38. Briefly, 6.0 g of phenol was mixed with 1.0 g of 20 wt% NaOH aqueous solution under stirring, and 10.0 g of formaldehyde solution was then added. The resulting transparent solution was stirred at 75 °C for 1 h, cooled to room temperature, and the pH was then adjusted to about 7.0 by 2.0 mol L−1 HCl. After water was removed by distillation, the mixture was redispersed in a required amount of ethanol. The NaCl precipitate was removed by filtration, and the filtrate, the P-F resol precursor with a concentration of 50 wt% in ethanol, was collected for further use. For a typical synthesis of manganese precursor (MP), 0.23 g of Mn(NO3)2 solution (50 wt%) was dissolved in 10.0 g of ethanol under stirring for 30 min. Next, 1.0 g of P-F resol was added to the solution and stirred for another 1 h. The above resulting mixture was added into 0.2 g of F127 and then stirred overnight.Preparation of MnOx/3DOMC catalyst
The MnOx/3DOMC was prepared by employing the CCTS with the as-prepared MP. Typically, the PMMA template was firstly soaked in the MP for 30 min. Care was taken to keep the solution level below the top of the PMMA template. After wiping off the excess solution, the infiltered template was dried at 100 °C for 2 h. Then, the soaking-drying steps were repeated for three times to ensure a complete filling of the void spaces between the PMMA microspheres. Afterwards, the product was pyrolyzed under flowing N2 at 450 °C for 2 h and then at 800 °C for another 3 h with a heating rate of 1 °C min−1 to remove the PMMA template and carbonize P-F resol. Finally, the desired MnOx/3DOMC catalyst was obtained. The total loading amount of manganese in the nanocomposite was 7.1 wt%, which was determined by inductively coupled plasma (ICP) analysis.For comparison, commercial Norit activated carbon (NAC) and TiO2 were used as supports for preparing the MnOx/NAC and MnOx/TiO2 catalysts by a conventional impregnation method. Briefly, 0.2 g of NAC or TiO2 was dispersed in a certain amount of Mn(NO3)2 solution (50 wt%). The mixture was ultrasonicated for 1 h and then dried overnight at 80 °C. Then, the catalysts were calcined under flowing N2 at 500 °C for 3 h. The total loading amounts of manganese on MnOx/NAC and MnOx/TiO2 were 7.3 wt% and 7.4 wt% (ICP analysis), respectively.
Characterizations
Scanning electron microscope (SEM) experiments were performed with a JSM 6360-LV electron microscope. The samples were vapor-deposited with gold before observation. X-ray diffraction (XRD) patterns were collected with a D/Max-βb diffractometer using a Cu Kα radiation source (λ = 0.15432 nm). Wide-angle diffractions were recorded at a scanning speed of 5° min−1. X-ray photoelectron spectroscopy (XPS) measurements were conducted using a PHI 5700 ESCA spectrometer with amonochromated Al Kα radiation (hν = 1486.6 eV). Spectra correction was conducted using the C 1s line at 284.6 eV. Chemical compositions of the samples were determined by ICP spectrometer (ICP-AES) on an IRIS Intrepid II XSP instrument. Transmission electron microscopy (TEM) images were obtained on a JEOL 2000EX electron microscope operating at an accelerating voltage of 120 kV. Prior to the observation, the samples were ultrasonically dispersed in ethanol and then dropped onto carbon-coated copper grids. Nitrogen adsorption–desorption isotherms were measured at −196 °C on a Micromeritics ASAP 2010 apparatus. The specific surface areas of samples were calculated by the Brunauer–Emmett–Teller (BET) equation. The total pore volumes (Vp) were estimated from the adsorption branches at a relative pressure (P/P0) of 0.995. The mesopore distributions and the mesopore volumes (Vmeso) were derived from the desorption branches of the isotherms using the BJH method while the micropore volumes (Vmic) were obtained by the t-plot method. Temperature-programmed desorption measurements of ammonia (NH3-TPD) of the catalysts were performed with a FineSorb 3010p NH3-TPD automated catalyst characterization apparatus. 0.10 g of catalyst was loaded in a quartz reactor, pretreated at 200 °C for 1 h in He flow (20 mL min−1), and cooled down to 100 °C ready for measurement. The adsorption of NH3 was performed at 100 °C using pulse model. After adsorption saturation, the catalyst was heated linearly with a rate of 10 °C min−1, from 100 to 700 °C under a constant He flow of 30 mL min−1. The desorbed amount of NH3 was monitored with a thermal conductivity detector (TCD). Diffuse reflectance infrared Fourier transform spectrum (DRIFTS) was measured on a Bruker Tensor 27 Fourier transform infrared spectrometer (FTIR) equipped with a heated and evacuated custom-made reaction cell with CaF2 windows connected to a gas-dosing and evacuation system. The catalyst was pressed into a self-supporting wafer with a diameter of 20 mm and a weight of 50 mg. Prior to acquisition of the DRIFTS, the catalyst was pretreated at 200 °C for 1 h in He flow (20 mL min−1) to clean the catalyst surface. After cooling to the room temperature and collecting the background spectra, the catalyst was exposed to pyridine steam for 30 min. Then the reaction cell was evacuated to remove weakly physisorbed pyridine, and the DRIFTS was recorded at room temperature with a spectra resolution of 4 cm−1. Temperature-programmed reduction of hydrogen (H2-TPR) of the catalysts were carried out on an AutoChem II 2910 chemisorber using 5% H2 diluted with N2 (50 mL min−1). 0.1 g of catalyst was firstly pretreated under N2 flow at 200 °C for 1 h. After cooling to room temperature, the catalyst was programmed heated to 700 °C at a ramping rate of 10 °C min−1. The H2 consumption during the experiment was monitored by a TCD.Catalytic measurements
The catalytic activities of the catalysts for the NH3-SCR were carried out in a quartz tube fixed-bed continuous flow microreactor containing 200 mg of the sieved catalyst (40–60 mesh). The typical feed gas composition was as follows: 1000 ppm of NO, 1000 ppm of NH3, 5% of O2, 5% of water vapor (if necessary), 200 ppm of SO2 (if necessary), and the balance of He. The water vapor was generated by passing He flow through a gas-wash device containing de-ionized water. The total flow rate was 300 mL min−1, which was equivalent to a gas hourly space velocity (GHSV) of 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 for the MnOx/3DOMC and MnOx/NAC catalysts, and 40000 h−1 for the MnOx/TiO2 catalyst (based on their actual volumes). The molar flow rate of the NO in feed gas was 2.23 × 10−7 mol s−1. The concentration of NO in the inlet and outlet gas was measured by a NOx analyzer (Eco Physics CLD 70S). All of the data were collected after 20 min when the SCR reaction reached a steady state. The NO conversion is calculated from the variation of the detected NO concentration: NO conversion (%) = (1 − [NO]out/[NO]in) × 100, where [NO]in is the inlet NO concentration and [NO]out is the outlet NO concentration. Preliminary testing showed that the current MnOx/3DOMC catalyst possessed the most intact-solid 3DOM architecture with a relatively high manganese content and the best NH3-SCR activity. Decreasing or further increasing the amount of Mn(NO3)2 in the MP caused pores blockage and damage of the corresponding product, and thus was disadvantageous to the increase of the NH3-SCR activity (Fig. S1 and S2†).
000 h−1 for the MnOx/3DOMC and MnOx/NAC catalysts, and 40000 h−1 for the MnOx/TiO2 catalyst (based on their actual volumes). The molar flow rate of the NO in feed gas was 2.23 × 10−7 mol s−1. The concentration of NO in the inlet and outlet gas was measured by a NOx analyzer (Eco Physics CLD 70S). All of the data were collected after 20 min when the SCR reaction reached a steady state. The NO conversion is calculated from the variation of the detected NO concentration: NO conversion (%) = (1 − [NO]out/[NO]in) × 100, where [NO]in is the inlet NO concentration and [NO]out is the outlet NO concentration. Preliminary testing showed that the current MnOx/3DOMC catalyst possessed the most intact-solid 3DOM architecture with a relatively high manganese content and the best NH3-SCR activity. Decreasing or further increasing the amount of Mn(NO3)2 in the MP caused pores blockage and damage of the corresponding product, and thus was disadvantageous to the increase of the NH3-SCR activity (Fig. S1 and S2†).
Calculation of turnover frequencies (TOFs)
The TOF, reflecting the conventional calculation of turnover frequency of NO over each manganese atom, is investigated in order to compare the intrinsic activities of the present MnOx/3DOMC catalyst with other MnOx-based catalysts previously reported in the NH3-SCR of NOx. Since the active phase of catalyst, MnOx, is not atomically dispersed on the support, the relative TOF of NO over per Mn atom is defined as the following equation:39where XNO is the NO conversion at certain temperature, VNO is the flow rate of NO (mol s−1), mCat is the amount of catalyst (g), X is the manganese content in the catalyst determined from XPS (wt%), and M is the molar weight of manganese (54.9 g mol−1).

Results and discussion
Characteristics of MnOx/3DOMC
Fig. 1a depicts the representative SEM image of the PMMA template. It was seen that the PMMA template was perfectly uniform and orderly with long range in a large area. A face-centered cubic array of microspheres of ∼320 nm with different facets could be clearly observed from the fractured surface. Fig. 1b depicts the representative SEM image of the MnOx/3DOMC catalyst. It was observed that the MnOx/3DOMC displayed a high-quality 3DOM structure with a small quantity of point defects and line defects. The macropore size and wall thickness of the sample were ∼190 nm and ∼50 nm, respectively, which corresponded to shrinkage of ∼40% compared with the initial size of the PMMA microspheres. In addition, the next layer was highly visible through the defects, and the voids were interconnected through open window. | ||
| Fig. 1 SEM images of: (a) the PMMA template; (b) the MnOx/3DOMC catalyst. | ||
XRD results of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts are compared in Fig. 2. For the MnOx/3DOMC, only two broad diffraction peaks at 22.5° and 43.5° characteristic of amorphous carbon appeared,40,41 and no any crystalline phases of MnOx were observed, suggesting their possible high dispersion in the 3DOM carbon matrices. For the MnOx/NAC, in addition to the diffraction peaks of amorphous carbon, two diffraction peaks at 43.2° and 50.5°, ascribed to the characteristics diffraction peaks of tetragonal symmetry of α-MnO2 (JCPDS 44-0141), were observed clearly, indicating the presence of MnOx with larger particle sizes in this catalyst than in the MnOx/3DOMC. In addition, no diffraction peaks for other manganese oxides were detected. For the MnOx/TiO2, the different diffraction peaks, assignable to the anatase TiO2 (JCPDS 21-1272), MnO2 (JCPDS 44-0141) and Mn2O3 (JCPDS 41-1442), respectively, could be found easily.
 | ||
| Fig. 2 XRD patterns of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. | ||
Fig. 3 presents the N2 adsorption–desorption isotherms and pore size distributions (PSDs) of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. For the MnOx/3DOMC, the isotherm belonged to a mixed type combining macropores with certain meso- and micropores, which exhibited a type II isotherm with an H3-type hysteresis loop and three N2 uptake phases. Below the very low relative pressure of P/P0 = 0.1, there was a significant quick rise of adsorption branch, indicating the presence of some micropores generated possibly from the carbonization of P-F resol. With the increase of the relative pressure from P/P0 = 0.1 to P/P0 = 0.8, the N2 adsorption amounts increased gradually accompanying with a slight H2-type hysteresis loop related to the capillary condensation taking place, implying the existence of some mesopores of a variety of sizes. At high relative pressures of P/P0 = 0.8–1.0, an obvious H3-type hysteresis loop, which had no clear adsorption plateau at P/P0 ≈ 1.0, indicative of a macroporous size distribution, was observed. Such a phenomenon was also observed in the others surfactants-assisted preparation of 3DOM materials,42–49 feature of combination of macro-mesopore structure. The PSD of the MnOx/3DOMC, which centered at ∼5 nm and ∼60 nm, respectively, also confirmed this point. In comparison with the MnOx/3DOMC, the hysteresis loops of the MnOx/NAC and MnOx/TiO2 at the low and high relative pressure ranges varied from sample to sample, reflecting their discrepancy in PSD with the MnOx/3DOMC. For the MnOx/NAC, three peaks centering at ∼3.0, 4.1, and 6.5 nm were observed in the PSD curve. Moreover, broad peaks between 20–60 nm were also observed, implying the inhomogeneity of its PSD. For the MnOx/TiO2, a wide PSD from 20 to 300 nm with two small bimodal distributions centering at ∼30 and ∼110 nm was observed, which possibly originated from the interstitial spaces between agglomerated particles. Table 1 lists the corresponding textural properties of the three catalysts. The MnOx/NAC possessed a BET surface area of 659 m2 g−1 and a Vp of 0.38 cm3 g−1. Its Vmeso and Vmic were 0.10 and 0.26 cm3 g−1, respectively, indicating that substantial micropores were present and contributed to the major surface area of the catalyst. On the contrary, despite the fact that the MnOx/3DOMC had a similar BET surface area of 656 m2 g−1 to the MnOx/NAC, the Vmeso strikingly increased to 0.36 cm3 g−1 accompanying with a distinct decrease of corresponding Vmic to 0.04 cm3 g−1, suggesting that the introduction of F127 during the preparation process could substantially increase the mesoporosity, which favored the significant enhancement in surface area of the obtained MnOx/3DOMC catalyst. In addition, the MnOx/TiO2 had the smallest the BET surface area of 68 m2 g−1 and the pore volume of 0.11 cm3 g−1 among the catalysts.
 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms and corresponding pore size distributions of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. | ||
| Catalyst | SBET (m2 g−1) | Vmeso (cm3 g−1) | Vmicro (cm3 g−1) | Vp (cm3 g−1) | Mn content (wt%) |
|---|---|---|---|---|---|
| MnOx/3DOMC | 656 | 0.36 | 0.04 | 0.43 | 7.1 |
| MnOx/NAC | 659 | 0.10 | 0.26 | 0.38 | 7.3 |
| MnOx/TiO2 | 68 | 0.05 | 0.01 | 0.11 | 7.4 |
Since the incomplete certain of manganese valence, XPS was adopted to further identify the components, oxidation states of manganese, and corresponding surface atomic concentration information of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts, as shown in Fig. 4. For the MnOx/3DOMC, the binding energies (BEs) of Mn 2p3/2 and 2p1/2 located at 642.1 and 653.8 eV, respectively, with a spin-energy separation of 11.7 eV, suggesting that the predominant oxidation state of manganese is +4.50 For the MnOx/NAC, the BEs of Mn 2p3/2 and 2p1/2 located at about 642.0 and 653.9 eV, respectively, which can be considered to be identical to those of the MnOx/3DOMC with a Mn4+ oxide phase. Unlike the MnOx/3DOMC and MnOx/NAC, the distinctive Mn 2p3/2 shoulder-peak of the MnOx/TiO2 could be deconvoluted into three characteristic peaks at BEs of 643.4 eV, 642.0 eV, and 640.3 eV, which were attributed to Mn4+, Mn3+, and Mn2+ oxide phases, respectively.51 Table 2 summarizes the surface atomic concentrations of manganese and the relative concentration ratios of Mn4+, calculated from the XPS spectra. The concentration ratio of Mn4+ over the MnOx/3DOMC, MnOx/NAC and MnOx/TiO2 catalysts were presented as 96%, 76%, and 49%. According to the total surface manganese concentrations over the catalysts, 1.65 at.% for the MnOx/3DOMC, 1.00 at.% for the MnOx/NAC, and 1.60 at.% for the MnOx/TiO2, the concentrations of Mn4+ over the catalysts were calculated to be 1.50 at.%, 0.76 at.% and 0.78 at.%, respectively. Evidently, the molar concentration of Mn4+ on the MnOx/3DOMC was much higher than that on the other two catalysts. Such discrepancy was presumably associated with the difference in catalyst preparation. In our case, the MnOx/3DOMC catalyst prepared by the dual-CCTS possessed higher manganese precursor (Mn(NO3)2) dispersion and exposed more manganese atoms on the surface than the other two catalysts prepared by the impregnation method. During the pyrolysis process, these exposed active atoms were thus easier to decompose directly into the high valence. Furthermore, the well developed interconnected networks of the ordered macro-mesopore structures of the 3DOM carbon would potentially afford much more internal surface to expose the active atoms. It was much reported that Mn4+ species and their redox processes were responsible for the high activity over the manganese-based catalysts in the low temperature NH3-SCR reaction. And a high Mn4+ ratio would enhance the oxidation of NO to NO2, which was beneficial to promote the low-temperature SCR activity.17,52–55 Therefore, the higher atomic concentration of Mn4+ would play an important role in obtaining better low-temperature NOx removal ability for the MnOx/3DOMC catalyst than for the other two catalysts. On the other hand, the O 1s spectrum is frequently used to identify the types of surface oxygen species in a particular oxide. The chemical environment of oxygen in metal oxide catalysts often has an important effect on their catalytic properties. Fig. 4 also shows the O 1s spectra of the three catalysts. For all the catalysts, three deconvoluted peaks were observed, which were attributed to three types of oxygen species: the peak at BE of 529–530 eV was assigned to the lattice oxygen O2− in Mn–O–Mn (denoted as Oβ), the peak at BE of 531–532 eV was ascribed to the surface oxygen ions with low coordination (denoted as Oα), such as O− or O22− belonging to the hydroxide (Mn–OH) or defective oxides, and the peak at a higher BE of above 533 eV corresponded to adsorbed water.53,56–59 It was established that the Oα were more active than the Oβ due to its higher mobility than the Oβ.60 As a result, the higher Oα/(Oα + Oβ) ratio was favorable to the NH3-SCR reaction owing to the enhanced oxidation of NO to NO2. Based on the XPS, the concentrations of Oα/(Oα + Oβ) over the MnO2/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts were calculated to be 14.1%, 3.8% and 10.1%, respectively (Table 2). Considering these results, as well as the manganese valence analysis, it was therefore reasonable to expect that the MnOx/3DOMC catalyst would potentially achieve better activity than the other two catalysts in the NH3-SCR reaction at low temperature.
 | ||
| Fig. 4 XPS spectra for: Mn 2p and O 1s of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. | ||
| Catalyst | Mn4+/(Mn2+ + Mn3+ + Mn4+) (%) | Mn (at.%) | Mn4+ (at.%) | Mn (wt%) | Oα/(Oα + Oβ) (%) | O (at.%) | Oα (at.%) |
|---|---|---|---|---|---|---|---|
| MnOx/3DOMC | 91 | 1.65 | 1.50 | 6.8 | 80.4 | 17.55 | 14.1 |
| MnOx/NAC | 76 | 1.00 | 0.76 | 4.2 | 29.3 | 13.10 | 3.8 |
| MnOx/TiO2 | 49 | 1.60 | 0.78 | 2.5 | 26.1 | 38.6 | 10.1 |
Fig. 5 illustrates typical TEM images of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. It was clearly seen in Fig. 5a that the MnOx/3DOMC possessed a high-quality 3DOM structure containing interconnected networks with overlapped pores. The macroporous diameter of the MnOx/3DOMC was ∼200 nm, in good agreement with its SEM observation. A clearer observation in the partial magnified image in Fig. 5b revealed that MnOx nanoparticles were highly distributed on the macropore walls of the 3DOM carbon scaffold. The sizes of MnOx nanoparticles fell in the range of 2–4 nm, as seen by the tiny black spots. From the high-resolution TEM image in Fig. 5c, the high dispersion MnOx on 3DOM carbon was much easier to be observed. Two crystal lattice fringes with a d-spacing of 0.239 and 0.418 nm corresponding to the (110) and (211) planes of MnO2 respectively can be clearly observed as well, whereas no significant crystalline plane of carbon was found, substantiating its amorphous nature. In addition, there were numerous mesopores with an average size of 5 nm randomly distributed in the macropore walls of the 3DOM carbon scaffold. The emergence of the mesopores could potentially enhance the capability of absorption and activation for gas reactant. Similar formation pathways have been discussed by other groups.42,61 Differing greatly from the MnOx/3DOMC, the TEM image in Fig. 5d displayed a relatively low dispersion of the MnOx particles with sizes of 20–100 nm in the MnOx/NAC. Similarly for the MnOx/TiO2, as shown in Fig. 5e, a large number of aggregates were formed.
 | ||
| Fig. 5 TEM images of: (a) (b) the MnOx/3DOMC catalyst at low and high magnification; (c) the MnOx/NAC catalyst; (d) the MnOx/TiO2 catalyst. | ||
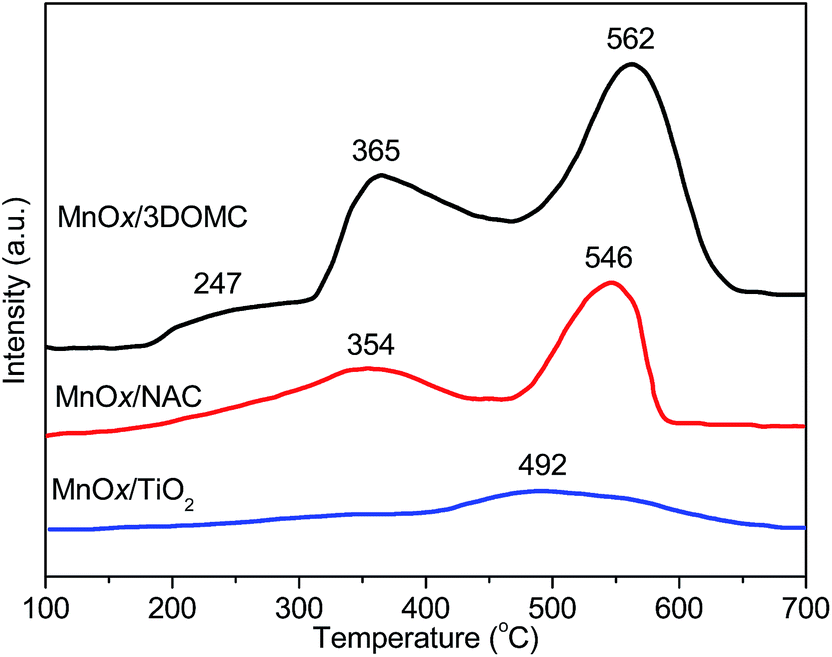
The NH3-TPD technique is often employed to determine the surface acid amount and strength of catalysts. The area of desorption peak is directly proportional to the acid amount and the peak position is correlated with the acid strength. Fig. 6 shows the NH3-TPD profiles of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. For the MnOx/3DOMC, the NH3 desorption profile exhibited three distinct desorption peaks centered at 247 °C, 365 °C, and 562 °C, assignable to the NH3 desorbed by weak, medium and strong acid sites, respectively.62,63 It was well accepted that the coordinated NH3 molecular bound to the Lewis (L) acid sites was more thermally stable than the NH4+ ions fixed on the Brønsted (B) acid sites.64 It was therefore conjectured that the desorption peak at low temperature was assigned to physisorbed NH4+ ions bound to the B acid sites, while the desorption peak at high temperature was associated with NH3 molecular originating from the L acid sites.65 On the other hand, pyridine is commonly used as a basic probe molecule for characterization of both B and L acidic sites by FTIR spectroscopy. In order to ascertain the type of B or L acid, we further characterized the MnOx/3DOMC by pyridine-DRIFTS. As shown in Fig. S3,† the band at 1446 cm−1 was typically attributed to pyridine bound to L acid sites, the band at 1487 cm−1 was assigned to the L or B acid sites, and the band at 1540 cm−1 was generally assigned to pyridine bound to B acid sites.66,67 Evidently, the strong and medium L acid sites were the main acid sites in the MnOx/3DOMC, whereas the peak belonging to B acid sites was relatively weak, indicating that only a small number of B acid sites were formed. For the MnOx/NAC, there were two major NH3 desorption peaks centered at 354 °C and 546 °C. Moreover, the overall curve shifted towards the low temperature region, indicating that the strength of acid sites on MnOx/NAC became weaker than that on MnOx/3DOMC. Meanwhile, the smaller area of desorption peaks of MnOx/NAC also implied a lower number of acid sites on this catalyst. For the MnOx/TiO2, the NH3 desorption peaks were relatively weak as compared with the other two samples, suggesting its shortage of acid sites. Such results indicated that the preparation method has a significant effect on the amount and the strength of the acidic sites on the catalysts. In our case, the MnOx/3DOMC was derived from P-F phenolic resin, which contained various aromatic structures supplying enough strong acid sites.68 On the other hand, it was reported that some oxides could increase the acidity of support remarkably.69 The largest area of the desorption peaks of the MnOx/3DOMC implied the most Brønsted and Lewis acid sites it possessed, which could be attributed to 3DOM carbon itself and the MnOx nanoparticles on 3DOM carbon.
 | ||
| Fig. 6 NH3-TPD profiles of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. | ||
H2-TPR is an ideal tool for investigating the reducibility of the MnOx over the different supports. Fig. 7 records the H2-TPR profiles of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. It was reported that the reduction process of MnOx took place in the following stepwise order: MnO2 → Mn2O3 → Mn3O4 → MnO.57,70 For the MnOx/3DOMC, there was one strong reduction peak centered at 233 °C, assigned to the reduction of major MnO2. For the MnOx/NAC, the reduction peak of MnO2 shifted to a higher temperature of 270 °C accompanying with the emergence of another obvious broad peak at 341 °C, which was correlated to the reduction of Mn2O3 phase. For the MnOx/TiO2, in addition to the peaks of the reduction of MnO2 to Mn2O3 (281 °C) and Mn2O3 to Mn3O4 (376 °C), the third peak at 464 °C, indicative of the reduction Mn3O4 to MnO, was observed. Apparently, the reduction temperature of MnO2 in the MnOx/3DOMC catalyst was found to be lower than those in the MnOx/NAC and MnOx/TiO2 catalysts, indicating the strongest reducibility of MnO2 in the MnOx/3DOMC catalyst. Furthermore, the larger area of the reduction peak of the MnOx/3DOMC catalyst meant the higher consumption of H2, suggesting that more active species were exposed in the MnOx/3DOMC catalyst than in the other two catalysts. This was in very good agreement with the TEM observation and XPS analysis.
 | ||
| Fig. 7 H2-TPR profiles of the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. | ||
NH3-SCR of NOx
Fig. 8 exhibits the plots of NO conversion percentage versus reaction temperature from 50 to 300 °C for the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. The NO conversion increased first and then decreased with increasing reaction temperature for all the catalysts. The catalytic activity of the MnOx/3DOMC was much higher than those of the MnOx/NAC and MnOx/TiO2. A nearly 100% NO conversion was achieved at 150 °C for the MnOx/3DOMC while it needed 210 °C for the MnOx/NAC. Compared with the MnOx/3DOMC and MnOx/NAC, the MnOx/TiO2 only completed 95% NO conversion at 230 °C. Evidently, the MnOx/3DOMC catalyst displayed the best catalytic performance. Based on the NO conversion at 100 °C, the TOF of the MnOx/3DOMC was calculated to be 3.81 × 10−4 s−1, which was higher than that of the MnOx/NAC (1.79 × 10−4 s−1) and MnOx/TiO2 (1.31 × 10−4 s−1). In addition, we also compared the TOF of the MnOx/3DOMC with some reported counterparts of MnOx-based NH3-SCR catalysts. As tabulated in Table 3, our result for the MnOx/3DOMC was much higher than those of the reported supported MnOx catalysts.11,14,17,71–79 The superior catalytic performance of the MnOx/3DOMC was mainly ascribed to the followings. The 3DOM carbon was the basic scaffold providing good accessibility for mass transport of the NO, NH3, and the corresponding product molecules, mechanical stability, and large surface area for absorbing reactants in the feed gas. The highly dispersed MnOx nanoparticles were directly introduced into carbon matrix, giving rise to more active sites and enabling the fast mass transfer for the reaction. In addition, the enhanced redox and acidic abilities of the MnOx/3DOMC played important roles in the NH3-SCR reaction. | ||
| Fig. 8 NO conversions over the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. Reaction conditions: 1000 ppm of NO, 1000 ppm of NH3, 5% of O2, He balance. | ||
| Catalyst | Mn content (wt%) by XPS | Composition of feed gas | GHSV (h−1) | Calculation temperature (°C) | TOF (s−1) | References | |
|---|---|---|---|---|---|---|---|
| NO + NH3 (ppm) | O2 (vol%) | ||||||
| MnOx/3DOMC | 6.8 | 1000 + 1000 | 5 | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 000 000 |
100 | 3.81 × 10−4 | This work |
| MnOx/NAC | 4.2 | 1000 + 1000 | 5 | 36000 |
100 | 1.79 × 10−4 | This work |
| MnOx/TiO2 | 2.5 | 1000 + 1000 | 5 | 40000 |
100 | 1.31 × 10−4 | This work |
| MnZ-3 | 16.76 | 600 + 600 | 4.5 | 36000 |
100 | 7.98 × 10−5 | 11 |
| MnOx/TiO2 | 5 | 500 + 500 | 2 | 75000 |
100 | 4.08 × 10−5 | 14 |
| MnOx/MWCNTs | 10 | 1000 + 1000 | 5 | 40000 |
100 | 2.57 × 10−4 | 17 |
| Mn-MA/Ti-CVC | 23.4 | 500 + 500 | 5 | 30000 |
100 | 4.89 × 10−6 | 71 |
| MnCBV | 3.5 | 500 + 500 | 5 | 80000 |
150 | 9.34 × 10−5 | 72 |
| MnOx/Al2O3–TiO2 | 8.2 | 500 + 500 | 5 | 80000 |
100 | 2.49 × 10−5 | 72 |
| MnOx/CZO | 25.74 | 1000 + 1000 | <5 | 6000 | 100 | 2.29 × 10−5 | 73 |
| MnOx/CeO2 | 7.22 | 600 + 660 | 6 | 45000 |
100 | 1.65 × 10−4 | 74 |
| MnOx/CeO2–ZrO2 | 9.4 | 600 + 600 | 3 | 30000 |
100 | 1.39 × 10−4 | 75 |
| MnOx/MWCNTs | 10 | 1000 + 1000 | 5 | 40000 |
100 | 5.90 × 10−5 | 76 |
| MnOx/CeO2 | 20 | 1000 + 1000 | 3 | 50000 |
100 | 2.04 × 10−4 | 77 |
| MnOx/CeO2–TiO2 | 20 | 200 + 200 | 8 | 60000 |
120 | 2.63 × 10−5 | 78 |
| (Ca)MnOx/TiO2 | 23.58 | 650 + 650 | 3 | 75000 |
100 | 7.39 × 10−5 | 79 |
Long-term stability is an important merit for catalysts. Meanwhile, since water vapor and/or SO2 are inevitable components of exhaust gas and may cause severe deactivation of catalysts for the NH3-SCR reaction, it is also necessary to study the water vapor resistance and/or SO2 tolerance of catalysts. Fig. 9 indicates the stability and the effects of water vapor and/or SO2 on NO conversion over the MnOx/3DOMC, MnOx/NAC, and MnOx/TiO2 catalysts. During a 48 h period at 190 °C (Fig. 9a), the MnOx/3DOMC preserved a high NO conversion at ∼98% with almost negligible activity decay. Unfortunately, the NO conversions over the MnOx/NAC and MnOx/TiO2 gradually faded with reaction time. After the continuous 48 h reaction, the 3DOM structure of the MnOx/3DOMC catalyst was still well-maintained with partial pore collapse to some extent, and the whole structural integrity remained considerably (Fig. 9e), implying that the long-term reaction did not spoil the structure of the catalyst and the high NO conversion could be further well preserved. The NH3-TPD profile after the long-term test shown in Fig. S4† also verified that there were indiscernible changes of the amount of acid sites and acid strength in the MnOx/3DOMC catalyst. When 5 v% water vapor was introduced into the feed gases (Fig. 9b), all the NO conversions over the three catalysts decreased during the test period. The fall range was ∼3.4% for MnOx/3DOMC, ∼5.0% for MnOx/NAC, and ∼9.3% for MnOx/TiO2, respectively, suggesting that the MnOx/3DOMC had a higher capacity for water vapor-resistance. After the water vapor was discontinued from the feed gases, all the NO conversions were almost restored to their originals, indicating that the inhibition effect of water vapor over the three catalysts was reversible. This was in good line with the reported results that the inhibition of water vapor resulted from the competitive adsorption between water and ammonia on the active sites of the catalyst's surface, which was reversible.80,81 When 200 ppm of SO2 was added to the feed gases (Fig. 9c), the NO conversion over the MnOx/3DOMC decreased slightly from initial 99% to 94%, then presented a gradually recovery trend during the test period. By contrast, the presence of SO2 caused a significant decrease of NO conversion over the MnOx/NAC and MnOx/TiO2 by 9 and 15%, respectively. After excluding SO2 in the feed gases, the conversions of NO over the MnOx/NAC and MnOx/TiO2 gradually restored to a certain extent, but still were less than their initial values, and finally returned to 81 and 58%, respectively. This substantiated that the MnOx/3DOMC had a stronger resistance to SO2. The NH3-TPD profile after the SO2-resistance test shown in Fig. S4† confirmed the remarkable increases in the amount of acid sites and acid strength in the MnOx/3DOMC catalyst. This was likely due to the increasing acidity of the catalyst by potential SO2 sulfuration, and implied that the acidity was not the only factor for the MnOx/3DOMC catalyst to determine the SCR performance. It was reported that the SO2 poisoning and deactivation to catalyst was usually related to the generation of ammonium sulfate species and deposition on the catalyst surface, blocking the active sites of the catalyst surface. And some MnOx-based catalysts resistance to SO2 could be enhanced due to the strong interaction between MnOx and the support with the inhibition of manganese sulfate formation on catalyst surface.82,83 In our case, the present MnOx nanoparticles formed with the dual-CCTS were mainly imbedded in or anchored on the carbon walls, which would not only produce highly dispersed MnOx nanoparticles in the 3DOMC matrix, but it could also create intimate contacts and strong interactions between the MnOx nanoparticles and the carbon support. Based on this point, such unique feature of the MnOx/3DOMC obviously helped to improve its SO2 resistance ability. In addition, the effect of coexistence of water vapor and SO2 was investigated. When 5 v% water vapor and 200 ppm SO2 were together introduced into the feed gases (Fig. 9d), the variation of NO conversion of the three catalysts were similar as those in the SO2-existence model. All the NO conversions exhibited a trend of first decease, then gradual recovery to a certain extent but less than their initial values, and finally returned to some stable values during the test period. This was likely stemmed from the synergistic inhibition effect of water vapor and SO2.
 | ||
| Fig. 9 (a–d) Stability tests and effect(s) of water vapor (and/or SO2) on NO conversion over the MnOx/3DOMC (■), MnOx/NAC (●), and MnOx/TiO2 (▲) catalysts. Reaction conditions: 190 °C, 1000 ppm of NO, 1000 ppm of NH3, 5% of O2, 5% of water vapor, and/or 200 ppm of SO2, He balance. (e) SEM image of the MnOx/3DOMCcatalyst after completing 48 h reaction. | ||
The excellent catalytic performance of the MnOx/3DOMC catalyst was approximately described as the Fig. 10 demonstrating. In the macro aspect, the MnOx/3DOMC was hierarchically porous structure. There were numerous additional mesopores in the macropore walls of the well-developed interconnected monolithic MnOx/3DOMC framework. Moreover, the MnOx nanoparticles were highly dispersed in the macropore walls where intimate contacts and strong interactions between the MnOx nanoparticles and the carbon support could be well yielded. These features were favorable to provide easier mass transport and more accessible internal surface area for the reaction, increase the number of active sites, and prevent the MnOx nanoparticles aggregation and growth during reaction. Meanwhile in the microcosmic aspect, the NH3-SCR of NOx over the MnOx/3DOMC catalyst was possibly described as the following five underlying reactions (Fig. 10). Reaction (1) was the adsorption of gaseous ammonia on the acid sites (i.e. B acid sites and L acid sites) to form adsorbed ammonia species including ionic NH4+ and coordinated NH3. Reaction (2) was the activation of adsorbed ammonia species by Mn4+ to form amide species (–NH2). Reaction (3) was the oxidation of NO to NO2. Then, gaseous NO and NO2 were reduced by –NH2 on the surface to form N2 and H2O via reaction (4). Reaction (5) was the re-oxidization of formed Mn3+.
 | ||
| Fig. 10 The proposed reaction mechanism for NH3-SCR of NOx over the MnOx/3DOMC catalyst. | ||
Conclusions
We have successfully prepared the MnOx/3DOMC nanocomposite via a simple multi-component infiltration of 3D PMMA templates followed by a direct pyrolysis process. The integration of 3DOM carbon and MnOx enables such a nanocomposite to possess high MnOx dispersion and strong interaction between the two components. As a catalyst, the MnOx/3DOMC nanocomposite displayed better low-temperature NH3-SCR activity and a more extensive operating temperature window than the MnOx/NAC and MnOx/TiO2 catalysts prepared by a conventional impregnation method. In addition, the MnOx/3DOMC nanocomposite also presented favourable stability, water vapor, and/or SO2 resistance. Such excellent catalytic behavior can be attributed to the positive synergetic contributions of the MnOx/3DOMC nanocomposite, including easy mass transport, large accessible surface area, highly dispersed MnOx nanoparticles on the 3DOM carbon substrate, and the enhanced redox and acidic properties. These results should deepen the understanding of the role of combining hierarchical ordered macrostructure with MnOx in catalysis. Moreover, this work conceptually provides a way for tailoring/designing 3DOM metal (oxide)–carbon nanocomposites to many different catalysts for various catalytic reactions, and thus is worthy of further exploration.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21071072) and the Doctoral Program Foundation of Liaoning Province (no. 20091047). We thank Mr Gang Zhao (College of Environmental Science and Engineering, Dalian Maritime University) for running XPS measurements.Notes and references
- Z. M. Liu, J. H. Li and S. I. Woo, Energy Environ. Sci., 2012, 5, 8799–8814 CAS.
- F. Klingstedt, K. Arve, K. Eränen and D. Y. Murzin, Acc. Chem. Res., 2006, 39, 273–282 CrossRef CAS PubMed.
- P. Forzatti, I. Nova and E. Tronconi, Ind. Eng. Chem. Res., 2010, 49, 10386–10391 CrossRef CAS.
- P. G. W. A. Kompio, A. Brückner, F. Hipler, G. Auer, E. Löffler and W. Grünert, J. Catal., 2012, 286, 237–247 CrossRef CAS PubMed.
- P. G. Smirniotis, D. A. Peña and B. S. Uphade, Angew. Chem., Int. Ed., 2001, 40, 2479–2482 CrossRef CAS.
- G. S. Qi and R. T. Yang, Appl. Catal., B, 2003, 44, 217–225 CrossRef CAS.
- W. Tian, H. S. Yang, X. Y. Fan and X. B. Zhang, J. Hazard. Mater., 2011, 188, 105–109 CrossRef CAS PubMed.
- X. F. Tang, J. H. Li, L. Sun and J. M. Hao, Appl. Catal., B, 2010, 99, 156–162 CrossRef CAS PubMed.
- P. Maitarad, D. S. Zhang, R. H. Gao, L. Y. Shi, H. R. Li, L. Huang, T. Rungrotmongkol and J. P. Zhang, J. Phys. Chem. C, 2013, 117, 9999–10006 CAS.
- F. D. Liu, W. P. Shan, Z. H. Lian, L. J. Xie, W. W. Yang and H. He, Catal. Sci. Technol., 2013, 3, 2699–2707 CAS.
- X. R. Lou, P. F. Liu, J. Li, Z. Li and K. He, Appl. Surf. Sci., 2014, 307, 382–387 CrossRef CAS PubMed.
- Y. J. Kim, H. J. Kwon, I. S. Nam, J. W. Choung, J. K. Kil, H. J. Kim, M. S. Cha and G. K. Yeo, Catal. Today, 2011, 151, 244–250 CrossRef PubMed.
- J. L. Xie, D. Fang, F. He, J. F. Chen, Z. B. Fu and X. L. Chen, Catal. Commun., 2012, 28, 77–81 CrossRef CAS PubMed.
- S. J. Yang, Y. W. Fu, Y. Liao, S. C. Xiong, Z. Qu, N. Q. Yan and J. H. Li, Catal. Sci. Technol., 2014, 4, 224–232 CAS.
- M. Casapu, O. Kröcher and M. Elsener, Appl. Catal., B, 2009, 88, 413–419 CrossRef CAS PubMed.
- X. Tang, J. Hao, H. Yi and J. Li, Catal. Today, 2007, 126, 406–411 CrossRef CAS PubMed.
- L. S. Wang, B. C. Huang, Y. X. Su, G. Y. Zhou, K. L. Wang, H. C. Luo and D. Q. Ye, Chem. Eng. J., 2012, 192, 232–241 CrossRef CAS PubMed.
- X. Wang, Y. Y. Zheng, Z. Xu, X. L. Wang and X. P. Chen, RSC Adv., 2013, 3, 11539–11542 RSC.
- X. M. Wang, Y. N. Wang, L. Feng, P. G. Liu and X. Zhang, Chem. Eng. J., 2012, 203, 251–258 CrossRef CAS PubMed.
- X. Zhang, G. H. Li, H. Q. Zhang, X. M. Wang, J. Y. Qu, P. G. Liu and Y. N. Wang, Soft Matter, 2013, 9, 6159–6166 RSC.
- S. M. Sun, W. Z. Wang and L. Zhang, J. Mater. Chem., 2012, 22, 19244–19249 RSC.
- K. M. Ji, H. X. Dai, J. G. Deng, L. Y. Song, B. Z. Gao, Y. Wang and X. W. Li, Appl. Catal., B, 2013, 129, 539–548 CrossRef CAS PubMed.
- D. L. Ma, Z. Y. Cao, H. G. Wang, X. L. Huang, L. M. Wang and X. B. Zhang, Energy Environ. Sci., 2012, 5, 8538–8542 CAS.
- Z. Liu, J. H. Mi, Y. Yang, X. L. Tan and C. Lv, Electrochim. Acta, 2014, 115, 206–215 CrossRef CAS PubMed.
- Y. Chen, Y. J. Zhu and Z. G. Chen, Thin Solid Films, 2013, 539, 122–126 CrossRef CAS PubMed.
- M. A. Al-Daous and A. Stein, Chem. Mater., 2003, 15, 2638–2645 CrossRef CAS.
- J. Q. Zhao, P. Wan, J. Xiang, T. Tong, L. Dong, Z. N. Gao, X. Y. Shen and H. Tong, Microporous Mesoporous Mater., 2011, 138, 200–206 CrossRef CAS PubMed.
- A. Vu and A. Stein, Chem. Mater., 2011, 23, 3237–3245 CrossRef CAS.
- D. Z. Han, X. Li, L. Zhang, Y. H. Wang, Z. F. Yan and S. M. Liu, Microporous Mesoporous Mater., 2012, 158, 1–6 CrossRef CAS PubMed.
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC.
- A. Stein, B. E. Wilson and S. G. Rudisill, Chem. Soc. Rev., 2013, 42, 2763–2803 RSC.
- H. Arandiyan, H. X. Dai, J. G. Deng, Y. X. Liu, B. Y. Bai, Y. Wang, X. W. Li, S. H. Xie and J. H. Li, J. Catal., 2013, 307, 327–339 CrossRef CAS PubMed.
- N. D. Petkovich and A. Stein, Chem. Soc. Rev., 2013, 42, 3721–3739 RSC.
- H. J. Wan, B. S. Wu, H. W. Xiang and Y. W. Li, ACS Catal., 2012, 2, 1877–1883 CrossRef CAS.
- Y. Liu, W. Y. Yao, X. L. Cao, X. L. Weng, Y. Wang, H. Q. Wang and Z. B. Wu, Appl. Catal., B, 2014, 160–161, 684–691 CrossRef CAS PubMed.
- C. He, J. R. Li, X. Y. Zhang, L. Q. Yin, J. S. Chen and S. K. Gao, Chem. Eng. J., 2012, 180, 46–56 CrossRef CAS PubMed.
- H. N. Li, L. Zhang, H. X. Dai and H. He, Inorg. Chem., 2009, 48, 4421–4434 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, H. F. Yang, Z. Li, C. Z. Yu, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- C. Z. Sun, J. Zhu, Y. Y. Lv, L. Qi, B. Liu, F. Gao, K. Q. Sun, L. Dong and Y. Chen, Appl. Catal., B, 2011, 103, 206–220 CrossRef CAS PubMed.
- Z. Liu, A. Q. Wang, X. D. Wang and T. Zhang, Catal. Today, 2008, 137, 162–166 CrossRef CAS PubMed.
- Z. Liu, Y. Yang, J. H. Mi, X. L. Tan and Y. Song, Catal. Commun., 2012, 21, 58–62 CrossRef CAS PubMed.
- R. Z. Zhang, H. X. Dai, Y. C. Du, L. Zhang, J. G. Deng, Y. S. Xia, Z. X. Zhao, X. Meng and Y. X. Liu, Inorg. Chem., 2011, 50, 2534–2544 CrossRef CAS PubMed.
- H. Zhang, H. X. Dai, Y. X. Liu, J. G. Deng, L. Zhang and K. M. Ji, Mater. Chem. Phys., 2011, 129, 586–593 CrossRef CAS PubMed.
- Y. X. Liu, H. X. Dai, Y. C. Du, J. G. Deng, L. Zhang, Z. X. Zhao and C. T. Au, J. Catal., 2012, 287, 149–160 CrossRef CAS PubMed.
- Z. X. Zhao, H. X. Dai, J. G. Deng, Y. C. Du, Y. X. Liu and L. Zhang, Microporous Mesoporous Mater., 2012, 163, 131–139 CrossRef CAS PubMed.
- H. Arandiyan, H. X. Dai, J. G. Deng, Y. X. Liu, B. Y. Bai, Y. Wang, X. W. Li, S. H. Xie and J. H. Li, J. Catal., 2013, 307, 327–339 CrossRef CAS PubMed.
- K. M. Ji, H. X. Dai, J. G. Deng, H. Y. Jiang, L. Zhang, H. Zhang and Y. J. Cao, Chem. Eng. J., 2013, 214, 262–271 CrossRef CAS PubMed.
- Y. C. Wei, Z. Zhao, T. Li, J. Liu, A. J. Duan and G. Y. Jiang, Appl. Catal., B, 2014, 146, 57–70 CrossRef CAS PubMed.
- J. J. Xu, Z. L. Wang, D. Xu, F. Z. Meng and X. B. Zhang, Energy Environ. Sci., 2014, 7, 2213–2219 CAS.
- C. D. Wanger, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-ray Photoelectron Spectroscopy, PerkineElmer, Eden Prairie, 1978 Search PubMed.
- Z. H. Chen, Q. Yang, H. Li, X. H. Li, L. F. Wang and S. C. Tsang, J. Catal., 2010, 276, 56–65 CrossRef CAS PubMed.
- M. Kang, E. D. Park, J. M. Kim and J. E. Yie, Appl. Catal., A, 2007, 327, 261–269 CrossRef CAS PubMed.
- F. D. Liu, H. He, Y. Ding and C. B. Zhang, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS PubMed.
- B. Thirupathi and P. G. Smirniotis, J. Catal., 2012, 288, 74–83 CrossRef CAS PubMed.
- A. Sultana, M. Sasaki and H. Hamada, Catal. Today, 2012, 185, 284–289 CrossRef CAS PubMed.
- A. E. Fischer, K. A. Pettigrew, D. R. Rolison, R. M. Stroud and J. W. Long, Nano Lett., 2007, 7, 281–286 CrossRef CAS PubMed.
- P. R. Ettireddy, N. Ettireddy, S. Mamedov, P. Boolchand and P. G. Smirniotis, Appl. Catal., B, 2007, 76, 123–134 CrossRef CAS PubMed.
- Y. S. Wu, Y. X. Zhang, M. Liu and Z. C. Ma, Catal. Today, 2010, 153, 170–175 CrossRef CAS PubMed.
- E. Park, S. Chin, Y. S. Kim, G. N. Bae and J. Jurng, Powder Technol., 2013, 233, 131–136 CrossRef CAS PubMed.
- F. D. Liu and H. He, J. Phys. Chem. C, 2010, 114, 16929–16936 CAS.
- W. B. Sui, J. T. Zheng, Z. Yang and M. B. Wu, Mater. Lett., 2011, 65, 2534–2536 CrossRef CAS PubMed.
- L. Chmielarz, P. Kustrowski, M. Zbroja, B. Gil-Knap, J. Datka and R. Dziembaj, Appl. Catal., B, 2004, 53, 47–61 CrossRef CAS PubMed.
- Y. S. Shen and S. M. Zhu, Catal. Sci. Technol., 2012, 2, 1806–1810 CAS.
- R. B. Jin, Y. Liu, Z. B. Wu, H. Q. Wang and T. T. Gu, Chemosphere, 2010, 78, 1160–1166 CrossRef CAS PubMed.
- S. Roy, B. Viswanath, M. S. Hegde and G. Madras, J. Phys. Chem. C, 2008, 112, 6002–6012 CAS.
- J. A. Lercher, C. Grundling and G. E. Mirth, Catal. Today, 1996, 27, 353–376 CrossRef CAS.
- M. Akçay, Appl. Catal., A, 2005, 294, 156–160 CrossRef PubMed.
- C. Moreno-Castilla and F. J. Maldonado-Hódar, Carbon, 2005, 43, 455–465 CrossRef CAS PubMed.
- K. Keyvanloo, A. Mohamadalizadeh and J. Towfighi, Appl. Catal., A, 2012, 417–418, 53–58 CrossRef CAS PubMed.
- T. Mishra, P. Mohapatra and K. M. parida, Appl. Catal., B, 2008, 79, 279–285 CrossRef CAS PubMed.
- E. Park, S. Chin, J. Jeong and J. Jurng, Microporous Mesoporous Mater., 2012, 163, 96–101 CrossRef CAS PubMed.
- M. Stanciulescu, G. Caravaggio, A. Dobri, J. Moir, R. Burich, J. P. Charland and P. Bulsink, Appl. Catal., B, 2012, 123–124, 229–240 CrossRef CAS PubMed.
- J. H. Ko, S. H. Park, J. K. Jeon, S. S. Kim, S. C. Kim, J. M. Kim, D. Chang and Y. K. Park, Catal. Today, 2012, 185, 290–295 CrossRef CAS PubMed.
- B. X. Shen, X. P. Zhang, H. Q. Ma, Y. Yao and T. Liu, J. Environ. Sci., 2013, 25, 791–800 CrossRef CAS.
- B. X. Shen, Y. Y. Wang, F. M. Wang and T. Liu, Chem. Eng. J., 2014, 236, 171–180 CrossRef CAS PubMed.
- S. W. Pan, H. C. Luo, L. Li, Z. L. Wei and B. C. Huang, J. Mol. Catal. A: Chem., 2013, 377, 154–161 CrossRef CAS PubMed.
- L. Xu, X. S. Li, M. Crocker, Z. S. Zhang, A. M. Zhu and C. Shi, J. Mol. Catal. A: Chem., 2013, 378, 82–90 CrossRef CAS PubMed.
- S. M. Lee, K. H. Park and S. C. Hong, Chem. Eng. J., 2012, 195–196, 323–331 CrossRef CAS PubMed.
- T. T. Gu, R. B. Jin, Y. Liu, H. F. Liu, X. L. Weng and Z. B. Wu, Appl. Catal., B, 2013, 129, 30–38 CrossRef CAS PubMed.
- I. Giakoumelou, C. Fountzoula, C. Kordulis and S. Boghosian, J. Catal., 2006, 239, 1–12 CrossRef CAS PubMed.
- X. L. Mou, B. S. Zhang, Y. Li, L. D. Yao, X. J. Wei, D. S. Su and W. J. Shen, Angew. Chem., Int. Ed., 2012, 51, 2989–2993 CrossRef CAS PubMed.
- J. H. Huang, Z. Q. Tong, Y. Huang and J. F. Zhang, Appl. Catal., B, 2008, 78, 309–314 CrossRef CAS PubMed.
- Z. B. Wu, R. B. Jin, H. Q. Wang and Y. Liu, Catal. Commun., 2009, 10, 935–939 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16141f |
| This journal is © The Royal Society of Chemistry 2015 |