
Simultaneous chemiluminescence determination of citric acid and oxalic acid using multi-way partial least squares regression
Ali Mokhtari
*a,
Mohsen Keyvanfardb and
Iraj Emamic
aDepartment of Science, Golestan University, Gorgan, I. R. Iran. E-mail: alimo58@yahoo.com; Fax: +981732245964; Tel: +981732254164
bDepartment of Chemistry, Majlesi Branch, Islamic Azad University, Isfahan, I. R. Iran. E-mail: keyvan45638@yahoo.com; Tel: +983116272078
cDepartment of Physics, Isfahan University of Technology, Isfahan, I. R. Iran. E-mail: iraj_emami@yahoo.com; Tel: +989131047363
First published on 18th March 2015
Abstract
A novel kinetic chemiluminescence method has been proposed for the simultaneous determination of oxalic acid (OA) and citric acid (CA). The method is based on the catalytic effect of OA and CA in the chemiluminescence (CL) reaction of tris(1,10-phen)ruthenium(II) with Ce(IV). In the batch mode, OA gives a broad peak with the highest CL intensity at 0.7 second, whereas the maximum CL intensity of the CA appears at about 4.7 seconds after injection of Ce(IV) solution. Based on the differential rate of the CL reaction corresponding to CA and OA and different effect of Ce(IV) concentration on the CL intensity of these substances, a three dimensional data and multi-way partial least squares (N-PLS) regression method were developed for the simultaneous determination of CA and OA. After selecting the best operating parameters, calibration graphs were obtained over the concentration ranges 4.0 × 10−8–2 × 10−5 mol L−1 and 2.0 × 10−7–2.0 × 10−4 mol L−1 for OA and CA, respectively. The limits of detections were 2.0 × 10−8 mol L−1 for OA and 1.0 × 10−7 mol L−1 for CA. Relative standard deviation (RSD) of the method for 11 times simultaneous determination of 1.6 × 10−6 mol L−1 of OA and 3.2 × 10−6 mol L−1 of CA were 7.5% and 2.9%, respectively. The proposed method was successfully applied to the determination of the mixtures in synthetic samples, stain removers and anti-varroa mite formulations.
Ali Mokhtari received his BSc in chemistry from the Department of Chemistry of the Gilan University of Rasht, Iran, in 2001, his MSc in analytical chemistry from Department of Chemistry of the Isfahan University of Technology, Iran, in 2003, and his PhD from the Department of Chemistry of the Isfahan University of Technology, Iran, in 2009. He is assistant professor in analytical chemistry at the Golestan University, Gorgan, Iran. |
Introduction
There is a great worldwide demand for citric acid (CA) due to its low toxicity, mainly being used as an acidulant in the pharmaceutical and food industries.1 Other applications of CA can be found in detergents and cleaning products,2 cosmetics and toiletries.3 Oxalic acid (OA) is used in industry as a bleaching agent, radiator cleaner and spot and rust remover.4 OA is registered for use as a disinfectant to control bacteria and germs and also is used as an inert ingredient in pesticide formulations.5 OA and CA are present simultaneously in some pesticides,6 pharmaceuticals7 or cleaner formulations.2,8Some analytical techniques have been used for the simultaneous determination of OA and CA along with other organic acids, for example HPLC,4,9–13 capillary electrophorese14–18 and ion chromatography.19–21 However, there is not any report for the simultaneous chemiluminescence (CL) determination of these organic acids with or without using a separation technique up to now.
Most common cited advantages of CL reactions are the relatively simple instrumentation required, the low detection limits and wide dynamic ranges,22–24 having contributed to the interest in CL detection in HPLC25 and in flow injection analysis (FIA).26 CL is often described as a dark-field technique;23 because, it is usually measured in absence (or with low levels) of background light. This leads to very low detection limits compared to other optical techniques. However, some disadvantages are to be considered as well. The method suffers from the lack of selectivity.27 A CL reagent may yield significant emission not just for one unique analyte that leads to interference effects in methods without a separation stage. Moreover, CL emission intensities are sensitive to a variety of environmental factors such as temperature, solvent, ionic strength, pH, and other species present in the system.22,23
Several techniques have been suggested to increase the specificity of CL analysis; such as using masking agents,28 chromatography,29–34 and wavelength discrimination.35
Generally, simultaneous determination of compounds by CL methods, without using a separation technique, could be conducted by time resolved CL or chemometric-assisted methods. Ruiz et al.36 developed a stopped flow time-resolved CL method for the simultaneous determination of the binary mixtures of citrate and pyruvate. The method was based on the different rates of the CL reaction of these organic acids in Ru(bpy)32+–Ce(IV) CL system. The same reagents and method have also been used for the determination of oxalate–tartrate37 and pyruvate–tartrate mixtures.38 Pulgarín et al.39 also described a stopped flow technique for the simultaneous determination of morphine and naloxone in synthetic samples. In all of above mentioned CL methods, influence of sample matrix in selected times should be investigated for each component to ensure that the slope of the calibration curve of one analyte not affected by another.39 Therefore, only some given concentration ratio of analytes could be determined simultaneously in the mixture; because in some concentration ratios, peak of an analyte may be covered by another one.
Chemometric methods (generally partial least squares (PLS) algorithm) have also been used in CL methods for assisting in the simultaneous determination of analytes in mixture. For example, PLS has been used for the simultaneous determination of cobalt and copper,40 protocatechuic and caffeic acids,41 cobalt and chromium,42 cobalt and manganese,28 ascorbic acid and L-cysteine43 and morphine along with naloxone.44
In recent years, multi-way PLS (N-PLS)45 and support vector regressions46–48 were successfully used for simultaneous determination of binary mixtures in CL methods for the first time. N-PLS algorithm, which has been developed by R. Bro,49 maintains the three or higher dimensional structures of the data, with the capability of extracting more information of a kinetic system than the conventional two way PLS model.50
In quantitative analysis, a calibration set with data taken at increasing concentrations of the analytes is necessary. Therefore, concentration provides one dimension of the signal-concentration data array. To construct a three-way array, the other two dimensions can be provided by two-dimensional instrumentation including excitation-emission fluorimetric scans,51 or separation techniques coupled to UV-visible,52 infrared,53 or mass spectrometric detection.54,55 Another way of generating a two-dimensional data array is to follow a chemical reaction with an instrument providing uni-dimensional scans, e.g., a diode-array UV visible spectrophotometer.56 The kinetic-spectrophotometric information obtained, together with the multivariate calibration at several concentrations, gives rise to a three-way data array (three dimensional data) which can be useful to resolve mixtures of compounds with very similar properties.57,58
One of the main limitations for employing N-PLS in CL methods is attributed to the lack of the wavelength separation techniques in the common CL instruments. Therefore, unlike to spectrophotometric methods, wavelength couldn't be applied in most CL methods as a variable or discrimination factor. Consequently, obtaining a three-way array of data is relatively difficult for using in multi-way methods such as N-PLS.
In this work, we found that the impact of Ce(IV) concentration on the CL intensity of CA and OA is different. Therefore, a Ce(IV) concentration mode was added to the time and sample modes to obtain the three way (three dimensional) data. Analytical techniques coupled with a separation method, such as HPLC, capillary electrophorese and ion chromatography, provide multi-analyte information about related species, compounds and metabolites present in the sample. However, each of these methods often offers its own set of advantages and disadvantages. There are some disadvantages, such as several time-consuming manipulations, special training, or requirement of comparatively expensive equipment and they are not readily amenable to be cost-effective or to miniaturize instrumentation.59,60 Multi-way calibration methods play important roles in solving the problem of closely overlapping peaks. These methods utilize a mathematical separation procedure to substitute the traditional chemical separation procedure.61
The proposed method provides a simple analytical tool for the simultaneous determination of OA and CA without using a separation technique. This is attractive because it can reduce the use of more complex instrumental techniques and the cost of needed analytical equipment.
In this method, a batch mode was used for the simultaneous CL determination of OA and CA in an insecticide, a cleaning agent and synthetic samples, using N-PLS regression. In addition predictive ability of the N-PLS model has been compared with conventional PLS model.
N-PLS
The theoretical aspects of N-PLS method have been described in several books and reviews.49,50,62 In summary, multi-way regression method, N-PLS extends the traditional PLS algorithm to higher orders, using the multi-dimensional structure of the data for model building and prediction.49 In the case of three-way data, the model is given by the following equation:
 | (1) |
Experimental
Apparatus
CL analysis was applied using a 0.50 cm light path length quartz cell. The CL signal was measured with a CL analyzer with PMT (Hamamatsu, model R212) using a low pass filter whose output was connected to a data processing system with a PC. A schematic block diagram of the used instruments is shown in Fig. 1. | ||
| Fig. 1 Schematic block diagram of the CL instrument. | ||
Reagents
All the solutions were prepared using reagent grade chemicals and doubly distilled water. OA and CA standard solutions (1.0 × 10−2 mol L−1) were daily prepared by dissolving 0.1270 g of OA dihydrate (Sigma-Aldrich) and 0.2120 g of CA monohydrate (Sigma-Aldrich) in 100.0 mL volumetric flasks. Ru(II) solution (1.0 × 10−2 mol L−1) was prepared by dissolving 0.3640 g of dichlorotris (1,10-phen) ruthenium(II) hydrate (Sigma-Aldrich) in 50.0 mL water. Ce(IV) solutions were prepared by dissolving calculated amount of ceric ammonium nitrate (Riedel-de Haën) in 8.0 mL H2SO4 1.0 mol L−1 and diluting to the mark with distilled water in 100.0 mL volumetric flasks. In this way, Ce(IV) concentrations between 1.0 × 10−3 and 9.0 × 10−3 mol L−1 were prepared.Sample preparation
10 mL of anti-varroa mite solution was filtered through a 0.45 μm filter membrane before analysis. Then 1.0 mL of the filtrate was serially diluted with deionized water, by a factor of 5 × 104. 10 mL of cleaning agent (stain remover) was filtered through a 0.45 μm filter membrane before analysis. Then 1.0 mL of the filtrate was serially diluted with deionized water, by a factor of 1 × 104.For preparation of synthetic sample, 100.0 mL solution containing 10.0 mL ethanol, 0.5 g NaNO3, 0.5 g KCl and 0.5 g sucrose was prepared. Each time 1.0 mL of the synthetic sample along with the proper volume of OA and CA standard solutions were transferred into a 50 mL volumetric flask and the mixture was diluted to mark with water.
Calibration set
Based on our primary experiments, the CL intensity versus concentration was linear in the ranges 4.0 × 10−8 to 2.0 × 10−5 and 2.0 × 10−7 to 2.0 × 10−4 mol L−1 for OA and CA, respectively. In the linear range of each organic acid (OA and CA), four concentrations were selected and standard solutions including binary combination of substrates were prepared based on full factorial design (a design with all possible high/low combinations of all the input factors). By this choice of design, possible interactions and non-linearities can be accounted for.63 The N-PLS and PLS models were obtained using a total of 16 standard solutions, which were obtained by adding adequate volumes of OA and CA stock solutions into a 100.0 mL volumetric flask and dilution to the mark with water. Table 1 show the concentration matrix used in the calibration step.| Sample no. | OA (mol L−1) | CA (mol L−1) | Sample no. | OA (mol L−1) | CA (mol L−1) |
|---|---|---|---|---|---|
| 1 | 4.0 × 10−7 | 3.2 × 10−6 | 9 | 4.0 × 10−7 | 4.8 × 10−5 |
| 2 | 1.6 × 10−6 | 3.2 × 10−6 | 10 | 1.6 × 10−6 | 4.8 × 10−5 |
| 3 | 4.0 × 10−6 | 3.2 × 10−6 | 11 | 4.0 × 10−6 | 4.8 × 10−5 |
| 4 | 1.6 × 10−5 | 3.2 × 10−6 | 12 | 1.6 × 10−5 | 4.8 × 10−5 |
| 5 | 4.0 × 10−7 | 1.6 × 10−5 | 13 | 4.0 × 10−7 | 1.6 × 10−4 |
| 6 | 1.6 × 10−6 | 1.6 × 10−5 | 14 | 1.6 × 10−6 | 1.6 × 10−4 |
| 7 | 4.0 × 10−6 | 1.6 × 10−5 | 15 | 4.0 × 10−6 | 1.6 × 10−4 |
| 8 | 1.6 × 10−5 | 1.6 × 10−5 | 16 | 1.6 × 10−5 | 1.6 × 10−4 |
Experimental procedure
An aliquot (400 μL) of standard solution consisting of both organic acids along with 400 μL of 4.0 × 10−3 mol L−1 of Ru(phen)32+ were transferred into the 0.50 cm path light length quartz cell. Then, the cell was placed at its location in front of PMT and the program was started. After a few seconds, 200 μL acidic Ce(IV) was injected into the cell by a microsyringe and the peak-like CL emission was recorded by a computer for about 70 s (with interval times of 100 ms). Those data information were collected into Excel software.For constructing the three way data to use in N-PLS model, 600 points of time (equivalent to 60 s) from each peak (10 points before rising the peak and 590 points after rising the peak) were selected and reminder points were deleted. CL intensities of sixteen bi-component mixture solutions were recorded at 4 different concentrations of Ce(IV) including 0.001, 0.003, 0.005 and 0.007 mol L−1. In this way, a three-way data with dimensions of [16 × 4 × 600] was obtained.
Software
All computations were performed using Matlab (The Math. Works Inc., Natick, MA, USA) and the N-PLS analysis was carried out by using the N-way Toolbox for Matlab, freely accessible via Internet.64Results and discussion
Kinetic profile of CL reaction of Ru(phen)32+–acidic Ce(IV)-oxalic acid and/or citric acid
The methodology of the method is based on the fact that the reduction rates of OA and CA in the CL reaction of Ru(phen)32+ and acidic solution of Ce(IV), are different. The time required to reach maximum intensity is different for OA and CA. The former is 0.7 s whereas the latter is 4.7 s. The CL signal of OA is a sharp and intense peak whereas the CL signal of CA is a broad peak. Kinetic profiles of the acids are shown in Fig. 2. | ||
| Fig. 2 Kinetic profiles of (a) OA, (b) CA, (c) mixture of OA and CA. Conditions: OA: 4.0 × 10−6 mol L−1, CA: 1.1 × 10−4 mol L−1, Ce(IV): 0.005 mol L−1, Ru(phen)32+: 4.0 × 10−3 mol L−1. | ||
Influence of chemical variables
Concentration of Ce(IV) made a different effect on the CL intensity of OA or CA. At low concentrations of Ce(IV) (∼0.001 mol L−1), CL intensity of OA was more intense than that of CA. As Ce(IV) concentration was increased, the CL intensities of both compounds increased, but with different rates. As can be seen in Fig. 3, CL intensity of OA increased rapidly to 0.003 mol L−1 and then decreased to 0.009 mol L−1 Ce(IV). CL intensity of CA increased slowly to 0.003 mol L−1 Ce(IV) and it was increased to 0.007 mol L−1 with higher rates, then the CL intensity increased slowly with increasing Ce(IV) concentration to 0.009 mol L−1 Ce(IV). Ce(IV) concentration was used for constructing the three-way data as mentioned in experimental section.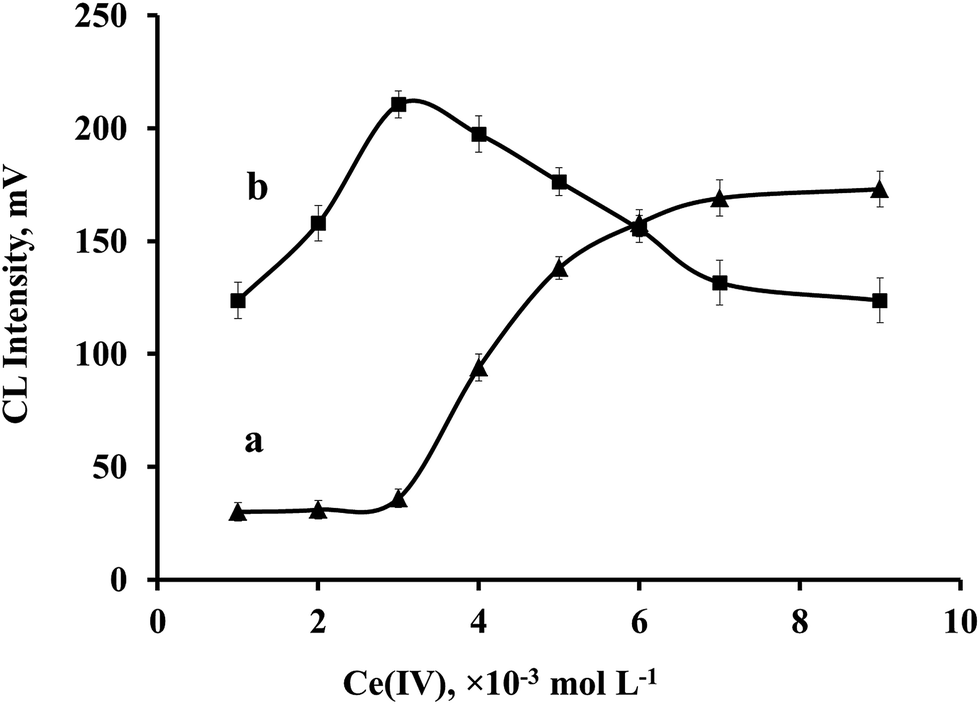 | ||
| Fig. 3 Influence of Ce(IV) concentration on the (a) CA and (b) OA CL intensities. OA: 4.0 × 10−6 mol L−1, CA: 1.1 × 10−4 mol L−1, Ru(phen)32+: 4.0 × 10−3 mol L−1. CL intensity: maximum value in the kinetic profile. | ||
The influence of concentration of H2SO4 on the CL intensity was studied in the range 0.04 to 0.16 mol L−1 of H2SO4. The CL response increased with increasing the concentration of H2SO4 to 0.08 mol L−1 and then decreased for both organic acids. Therefore, concentration 0.08 mol L−1 H2SO4 was selected for further studies.
The influence of concentration of Ru(phen)32+ on the sensitivity was also studied in the range 1.0 × 10−3 to 7.0 × 10−3 mol L−1 by injecting concentration of 5.0 × 10−3 mol L−1 of Ce(IV) prepared in 0.08 mol L−1 of H2SO4. The CL signal increased with increasing Ru(phen)32+ concentrations until 4.0 × 10−3 mol L−1 and then decreased for both OA and CA. Therefore, concentration of 4.0 × 10−3 mol L−1 was selected as the optimum concentration for the complex of Ru(phen)32+.
N-PLS regression
Since, the CL kinetic profiles of OA and CA are overlapped with each other a first or second order calibration is required to predict the concentration of each compound in the mixture. As it mentioned above, effect of Ce(IV) concentration in a definite range (0.001–0.009 mol L−1) had different influences on the CL intensity of the compounds. Therefore, it was thought that concentration of Ce(IV) has potential to be selected as a new variable for constructing a three-way data instead of working with two-way data. In this regard, a three-way data structure, [sample, Ce(IV) concentration, time], was constructed. The next step was selecting number of factors for each analyte using N-PLS regression. Number of factors and the performance of N-PLS model was evaluated by calculating the root mean squared errors of cross validation (RMSECV) for each analyte, which is defined as follows:65
 | (2) |
In that, yi is the reference concentration for the ith sample and ŷi represents the predicted concentration. In RMSECV method, one sample was eliminated at a time and then N-PLS model constructed with remaining standard samples. By using this calibration, the concentration of the sample, left out, was predicted. This value was calculated for different number of the factors in the model. The results are listed in Table 2. The optimum number of the factors was selected based on the minimum value for the RMSECV. It can be noticed that the RMSECV values are minimum for three and two factors for OA and CA, respectively. The number of the factors for OA is larger than the number of the analytes. This could be related to the non-linearity in the data, which could be compensated by enhancing the number of the factors in the model. Beyond the respective number of factors for OA and CA, the model was overfitted.
| Number of factors | |||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| RMSECV (OA, × 10−6) | 1.4 | 1.9 | 0.7 | 1.6 | 5.8 |
| RMSECV (CA, × 10−6) | 12.1 | 7.6 | 11.7 | 14.5 | 14.7 |
Analytical features
Under the optimum condition of each organic acid, a long series of standard solutions of OA and CA were separately subjected to the CL method for the purpose of calibration. CL response was found to be linear in the concentration ranges of 4.0 × 10−8 to 2 × 10−5 mol L−1 and 2.0 × 10−7 to 2.0 × 10−4 mol L−1 for OA and CA, respectively. Fig. 4 shows the calibration curves and respective linear equations for OA and CA. | ||
| Fig. 4 Calibration curves for OA and CA. CL intensity: maximum value in the kinetic profile. | ||
The limit of detection (LOD) was calculated as 3σ/m where σ is the standard deviation existing in 11 times determination of the blank response and m is slope of the calibration curve. LODs were 2.0 × 10−8 mol L−1 for OA and 1.0 × 10−7 mol L−1 for CA. The reproducibility was investigated and the percent of relative standard deviation (%RSD) for 3.2 × 10−6 mol L−1 of CA (n = 11) was 1.3%. RSD of the method also evaluated using N-PLS model. In this study, the CL responses obtained for 11 replications of the sample including 1.6 × 10−6 mol L−1 of OA and 3.2 × 10−6 mol L−1 of CA. for each replication, concentration of OA and CA predicted by the optimized N-PLS model. RSDs for OA and CA obtained 7.5% and 2.9%, respectively. The minimum sampling rate calculated about 20 samples per hour.
Influence of foreign compounds
To evaluate the selectivity of the proposed method, the influences of some common ingredients may be included in the cleaning agents and anti-mite (or pesticide) formulations and some other organic and inorganic substances on the determination of OA and CA were separately investigated. The tolerance of each substance was taken as the largest amount yielding an error of less than 3σ in the analytical signal of OA or CA (σ is the standard deviation in the response obtained from 11 times determination of 4.0 × 10−6 mol L−1 OA or 4.8 × 10−5 mol L−1 CA). In this study appeared that a 100-fold excess of sucrose, glucose, saccharine, lactose, fructose, ethanol, 2-ethylhexanol, K+, Cl−, Na+, NO3−, CN−, Br−, Zn2+, SO42−, Fe3+, PO43−, urea, isopropanol, phenol, diethylene glycol and NH4+, a 10-fold excess of I−, Ca2+, benzoic acid, borate, boric acid, CO32−, acetic acid and EDTA have no effect on the determination of OA (4.0 × 10−6 mol L−1) and CA (4.8 × 10−5 mol L−1). For tartaric acid the concentration must be below 0.1-fold to avoid interference with CA. In addition, to evaluate the selectivity of the proposed method, binary mixtures of both analytes along with some excipients such as lactose, sucrose, ethanol, 2-ethylhexanol, zinc sulphate, potassium nitrate, urea and sodium chloride were studied using N-PLS model. The procedure consisted of preparing different solutions with each one of these excipients with concentration of 1.0 × 10−3 mol L−1 and containing OA and CA at 4.0 × 10−6 mol L−1 and 4.8 × 10−5 mol L−1, respectively. The results are listed in Table 3.Application
In order to investigate the accuracy of the method, three samples including anti-varroa mite solution, stain remover solution and synthetic sample were analyzed to determine OA and CA contents. In this regard, samples were prepared as described in the experimental section and the predicted concentrations were obtained by the N-PLS model. The results are given in Table 4. The recoveries are in the range of 87 to 114%.| Added (×10−5 mol L−1) | Found (×10−5 mol L−1) | Recovery (%) | ||||
|---|---|---|---|---|---|---|
| CA | OA | CA | OA | CA | OA | |
| a Each 500 mL contains: CA 10 g, OA 15.5 g, ethanol 10 g. (Dany's BienenWohl, Austria).b Contains: CA 1% and OA 0.1%; (cleaning agent, Carbona Stain Devil no. 9, Delta pronatura-Germany). | ||||||
| Anti-varroa mitea | 0.00 | 0.000 | 0.22 | 0.671 | — | — |
| 1.00 | 0.500 | 1.21 | 1.206 | 99.0 | 107.0 | |
| 5.00 | 0.500 | 5.04 | 1.143 | 96.4 | 94.4 | |
| 5.00 | 0.100 | 5.75 | 0.766 | 110.6 | 95.0 | |
| 5.00 | 0.800 | 4.81 | 1.532 | 91.8 | 107.6 | |
| 10.00 | 0.500 | 10.43 | 1.108 | 102.1 | 87.4 | |
| Stain removerb | 0.00 | 0.000 | 0.46 | 0.127 | — | — |
| 1.00 | 0.500 | 1.34 | 0.642 | 93.6 | 103.0 | |
| 5.00 | 0.500 | 5.69 | 0.696 | 104.6 | 113.8 | |
| 5.00 | 0.100 | 5.16 | 0.228 | 94.0 | 101.0 | |
| 5.00 | 1.000 | 5.25 | 1.042 | 95.8 | 91.5 | |
| 10.00 | 0.500 | 10.59 | 0.652 | 101.3 | 105.0 | |
| Synthetic sample | 0.00 | 0.000 | 0.01 | 0.021 | — | — |
| 1.00 | 0.500 | 0.94 | 0.552 | 93 | 106.2 | |
| 5.00 | 0.500 | 4.94 | 0.504 | 98.6 | 96.6 | |
| 5.00 | 0.100 | 5.25 | 0.113 | 104.8 | 92.0 | |
| 5.00 | 1.000 | 4.87 | 1.123 | 97.2 | 110.2 | |
| 10.00 | 0.500 | 9.28 | 0.524 | 92.7 | 100.6 | |
In addition, prediction ability of the proposed method compared with a HPLC method for simultaneous determination of OA and CA in anti-varroa mite sample. In this study, the prediction results using N-PLS model were compared with results obtained by a HPLC method improved by Khaskhali et al.13 The results are shown in Table 5. It must be noticed that no replication and averaging has been performed in N-PLS method.
| Sample | Nominal value (g L−1) | Found (g L−1) | ||||
|---|---|---|---|---|---|---|
| Proposed method | Literature method13 | |||||
| OA | CA | OA | CA | OA | CA | |
| Anti-varroamite | 31 | 20 | 30.2 | 21.1 | 29.8 | 19.7 |
OA and CA are naturally-occurring substances. High oxalate in the urine and plasma was first found in people who were susceptible to kidney stones.66 CA is an important intermediate in metabolism. In humans, citrate is excreted by the kidney and it plays an important role as an inhibitor in preventing supersaturation with respect to the formation of calcium oxalate which is the most common constituent of kidney stones.13 The mechanism of inhibitory action of CA is probably through the chelating of Ca2+ ions in urine and thus, preventing the latter from combining with stone forming anions like oxalate.66 The low urinary citrate and significantly higher urinary oxalate levels may be a serious risk factor in calcium oxalate stone formation in kidney stone patients;13 therefore, citrate and oxalate determination has become an important tool in the assessment of urine supersaturation with respect to calcium oxalate. One future trend might be improving of the proposed CL method for simultaneous determination of citrate and oxalate in urine as a kidney stone diagnosis.
Comparison between PLS and N-PLS models
Among the chemometric methods, PLS algorithm, more than of other algorithms has been used in CL methods for assisting in the simultaneous determination of analytes in the mixtures.28,35,40–44 In order to compare results obtained by N-PLS with results from conventional two-way PLS, each time, one dimension of the calibration data was eliminated, in this way one slice of previous three-way data (applied for constructing N-PLS model) corresponding to one level of Ce(IV) concentration was selected. Therefore, the data including 16 samples × 600 times was employed for constructing PLS model. Next, number of factors was optimized for selected level of Ce(IV) concentration using RMSECV method for OA and CA as described in N-PLS regression section. Lowest amount of RMSECV was obtained for fourth level of Ce(IV) concentration (0.007 mol L−1) with five factors for both OA and CA. The predictive results of the PLS model at optimum number of factors and selected concentration of Ce(IV) was determined for anti-varroa mite sample (Table 6). In this manner previous 3D-data obtained for anti-varroa mite sample was converted to two dimensions matrix (one slice of data corresponding to 0.007 mol L−1 of Ce(IV) was selected and PLS model applied for simultaneous determination of OA and CA. As can be seen in Tables 4 and 6, no satisfactory recoveries could be obtained for OA and CA using conventional two-way PLS in compare to N-PLS model.| Added (×10−5 mol L−1) | Found (×10−5 mol L−1) | Recovery (%) | |||
|---|---|---|---|---|---|
| CA | OA | CA | OA | CA | OA |
| 0.00 | 0.000 | 0.230 | 1.240 | — | — |
| 1.00 | 0.500 | 1.293 | 1.378 | 106.3 | 27.6 |
| 5.00 | 0.500 | 5.536 | 1.743 | 106.1 | 100.6 |
| 5.00 | 0.100 | 3.946 | 1.350 | 74.3 | 110.0 |
| 5.00 | 0.800 | 15.425 | 1.727 | 303.9 | 60.9 |
| No. of factors | 5 | 5 | |||
Mechanism
Solution of Ru(phen)32+ is orange and its color changes to green immediate after mixing with oxidizing agent, Ce(IV) solution, and production of Ru(phen)33+.67,68 During about 3 minutes after mixing Ru(phen)32+ with Ce(IV), the color of the mixture changes slowly from green to orange. The resulting Ru(phen)33+ produced in the reaction of Ru(phen)32+ with acidic Ce(IV), is a powerful oxidant and oxidizes water into O2 and protons.69 Therefore, it returns slowly to its reduced state. If there was a reducing agent in the reaction media, it can reduce Ru(phen)33+ very fast. The electrons from reducing agent transfer to the π*-orbital of phenanthroline ligand and the Ru(phen)32+ π* metal-to-ligand charge transfer (MLCT) excited state can be produced.70 The excited electron then undergoes intersystem crossing to the lowest triplet state of Ru(phen)32+, from where emission occurs.71Ce(IV) is a one-electron oxidant and reacts with organic acids to form a reactive intermediate radical.72,73 The mechanism involves the rapid formation of an activated Ce(IV) complex followed by its slow decomposition (reactions 1–4).
These radical ions produce the excited state, [Ru(phen)32+]*, by an electron transfer reaction with trivalent ruthenium species (reaction 6). An emission having a maximum at 580 nm was produced when the excited state molecule of Ru(phen)32+ returns to the ground state.36
The kinetics of the oxidation of 16 organic acids including OA and CA by Ce(IV) have been investigated in the presence of Ru(phen)32+.74 It was found that all of the mentioned acids can form activated Ce(IV) complexes and produce radical anion which they can reduce the Ru(phen)33+ and enhance the CL emission. Therefore decomposition rate of reaction (4) is one of the factors which can determine the kinetic of the CL reaction. The time required to reach maximum CL intensity in the presence of Ce(IV) and Ru(phen)32+ is much shorter for OA than for CA. This suggests that the formation of the intermediate radical and its decomposition rate takes place at slower rates for CA. In addition, the reduction rate of Ru(phen)33+ to excited form, [Ru(phen)32+]*, by the intermediate radical is different for each acid.36 Based on the above mentions, a coupled CL mechanism of complexation and redox reactions is suggested.37 A detailed mechanism for the overall process is expressed as Scheme 1 (taking OA as an example).
 | ||
| Scheme 1 Detailed mechanism for the CL reaction of OA and CA. | ||
Conclusion
A CL method was introduced for the simultaneous determination of OA and CA using N-PLS regression. This paper demonstrates the usefulness of mathematical deconvolution of CL data by the N-PLS model from sample matrices as well as for peak purity evaluation in chromatography. The concentration of Ce(IV) was selected as one of the variables in the three-way data, because its influence on the CL intensity of OA and CA was different. The accuracy of the method was examined by analysis of the synthetic sample, stain remover and anti-varroa mite formulations. The results reveal the ability of the proposed method.Acknowledgements
We are grateful to the campus of Golestan University for supporting of this research.Notes and references
- S. Anastassiadis, I. G. Morgunov, S. V. Kamzolova and T. V. Finogenova, Recent Pat. Biotechnol., 2008, 2, 107–123 CrossRef CAS.
- A. D. Deshpande and B. B. Gogte, Res. J. Chem. Sci., 2011, 1, 42–47 CAS.
- C. R. Soccol, L. P. S. Vandenberghe, C. Rodrigues and A. Pandey, Food Technol. Biotechnol., 2006, 44, 141–149 CAS.
- M. O. Nisperos-Carriedo, B. S. Buslig and P. E. Shaw, J. Agric. Food Chem., 1992, 40, 1127–1130 CrossRef CAS.
- U.S. Environmental Protection Agency (EPA), Reregistration Eligibility Decision: Oxalic Acid, EPA 738-F92-014, 1992, http://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=P100AFN7.txt, accessed (February 2015).
- B. Dany, anti-varroa mite, Dany's Bienenwohl, Austria, http://www.bienenwohl.com/eng/bienenwohl.php, accessed (March 2014).
- C. Foret, A. Skender, F. Ahmed, T. C. Hemling and N. C. Traistaru, U.S. Pat. 8,569,373, 2013.
- Dr. Beckmann Stain Devils Rust & Deodorant No. 9, delta pronatura, Germany, 2009, https://www.carbona.com/MSDS/403%20-%20Carbona%20Stain%20Devils9.pdf, accessed (February 2015).
- Z. Chen and Y. Jin, J. Shaanxi Norm. Univ., Nat. Sci. Ed., 2006, 4, 14 Search PubMed.
- M. A. Kall and C. Andersen, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 730, 101–111 CrossRef CAS.
- E. Paredes, S. E. Maestre, S. Prats and J. L. Todolí, Anal. Chem., 2006, 78, 6774–6782 CrossRef CAS PubMed.
- C. Zhanguo and L. Jiuru, J. Chromatogr. Sci., 2002, 40, 35–39 Search PubMed.
- M. Hassan Khaskhali, M. Iqbal Bhanger and F. D. Khand, J. Chromatogr. B: Biomed. Sci. Appl., 1996, 675, 147–151 CrossRef.
- J. M. Izco, M. Tormo and R. Jiménez-Flores, J. Dairy Sci., 2002, 85, 2122–2129 CrossRef CAS.
- I. Mato, J. F. Huidobro, J. Simal-Lozano and M. T. Sancho, Anal. Chim. Acta, 2006, 565, 190–197 CrossRef CAS PubMed.
- M. Shirao, R. Furuta, S. Suzuki, H. Nakazawa, S. Fujita and T. Maruyama, J. Chromatogr. A, 1994, 680, 247–251 CrossRef CAS.
- J. Xu, Z. Chen, J. C. Yu and C. Tang, J. Chromatogr. A, 2002, 942, 289–294 CrossRef CAS.
- W. C. Yang, Y. Q. Dai, A. M. Yu and H. Y. Chen, J. Chromatogr. A, 2000, 867, 261–269 CrossRef CAS.
- Z. Chen and M. A. Adams, Anal. Chim. Acta, 1999, 386, 249–256 CrossRef CAS.
- F. Chinnici, U. Spinabelli, C. Riponi and A. Amati, J. Food Compos. Anal., 2005, 18, 121–130 CrossRef CAS PubMed.
- M. Y. Ding, Y. Suzuki and H. Koizumi, Analyst, 1995, 120, 1773–1777 RSC.
- W. Baeyens, S. Schulman, A. Calokerinos, Y. Zhao, A. M. G. Campana, K. Nakashima and D. De Keukeleire, J. Pharm. Biomed. Anal., 1998, 17, 941–953 CrossRef CAS.
- J. A. Ocaña-González, M. Ramos-Payán, R. Fernández-Torres, M. V. Navarro and M. Á. Bello-López, Talanta, 2014, 122, 214–222 CrossRef PubMed.
- Y. Hu and Z. Yang, Talanta, 2004, 63, 521–526 CrossRef CAS PubMed.
- H. Kodamatani, A. Matsuyama, K. Saito, Y. Kono, R. Kanzaki and T. Tomiyasu, Anal. Sci., 2012, 28, 959–965 CrossRef CAS.
- D. C. Christodouleas, D. L. Giokas, V. Garyfali, K. Papadopoulos and A. C. Calokerinos, Microchem. J., 2015, 118, 73–79 CrossRef CAS PubMed.
- T. A. Nieman, W. R. G. Baeyens, D. D. Keukeleire and K. Korkidis, Luminescence Techniques in Chemical and Biochemical Analysis, Marcel Dekker, New York, 1991 Search PubMed.
- Q. Lin, A. Guiraúm, R. Escobar and F. F. de la Rosa, Anal. Chim. Acta, 1993, 283, 379–385 CrossRef CAS.
- R. Hua, Y. Li, W. Liu, J. Zheng, H. Wei, J. Wang, X. Lu, H. Kong and G. Xu, J. Chromatogr. A, 2003, 1019, 101–109 CrossRef CAS PubMed.
- K. Nakagawa and T. Miyazawa, Anal. Biochem., 1997, 248, 41–49 CrossRef CAS PubMed.
- K. Nakashima, K. Suetsugu, S. Akiyama and M. Yoshida, J. Chromatogr. B: Biomed. Sci. Appl., 1990, 530, 154–159 CrossRef CAS.
- E. Nalewajko, A. Wiszowata and A. Kojło, J. Pharm. Biomed. Anal., 2007, 43, 1673–1681 CrossRef CAS PubMed.
- H. Wu, M. Chen, Y. Fan, F. Elsebaei and Y. Zhu, Talanta, 2012, 88, 222–229 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhang and Y. Sun, J. Chromatogr. A, 2006, 1129, 34–40 CrossRef CAS PubMed.
- A. S. Carretero, J. R. Fernandez, A. R. Bowie and P. J. Worsfold, Analyst, 2000, 125, 387–390 RSC.
- T. Pérez-Ruiz, C. Martínez-Lozano, V. Tomás and J. Fenoll, Anal. Chim. Acta, 2003, 485, 63–72 CrossRef.
- Z. He and H. Gao, Analyst, 1997, 122, 1343–1346 RSC.
- X. Li, L. Ling, Z. He, G. Song, S. Lu, L. Yuan and Y. E. Zeng, Microchem. J., 2000, 64, 9–13 CrossRef CAS.
- J. A. M. Pulgarín, L. F. G. Bermejo, J. M. L. Gallego and M. N. S. García, Talanta, 2008, 74, 1539–1546 CrossRef PubMed.
- B. Li, D. Wang, J. Lv and Z. Zhang, Talanta, 2006, 69, 160–165 CrossRef CAS PubMed.
- A. Navas Diaz and J. A. G. Garcia, Anal. Chem., 1994, 66, 988–993 CrossRef CAS.
- B. Li, D. Wang, J. Lv and Z. Zhang, Spectrochim. Acta, Part A, 2006, 65, 67–72 CrossRef PubMed.
- B. Li, D. Wang, C. Xu and Z. Zhang, Microchim. Acta, 2005, 149, 205–212 CrossRef CAS.
- J. A. Murillo Pulgarín, L. F. García Bermejo and M. N. Sánchez García, Anal. Chim. Acta, 2007, 602, 66–74 CrossRef PubMed.
- B. Rezaei, T. Khayamian and A. Mokhtari, J. Pharm. Biomed. Anal., 2009, 49, 234–239 CrossRef CAS PubMed.
- A. A. Ensafi, F. Hasanpour, T. Khayamian, A. Mokhtari and M. Taei, Spectrochim. Acta, Part A, 2010, 75, 867–871 CrossRef PubMed.
- A. A. Ensafi, F. Hasanpour and T. Khayamian, Talanta, 2009, 79, 534–538 CrossRef CAS PubMed.
- F. Hasanpour, A. A. Ensafi and T. Khayamian, Anal. Chim. Acta, 2010, 670, 44–50 CrossRef CAS PubMed.
- R. Bro, J. Chemom., 1996, 10, 47–61 CrossRef CAS.
- A. Smilde, R. Bro and P. Geladi, Multi-way analysis: applications in the chemical sciences, John Wiley & Sons, 2005 Search PubMed.
- M. G. Trevisan and R. J. Poppi, Anal. Chim. Acta, 2003, 493, 69–81 CrossRef CAS.
- E. Comas, R. A. Gimeno, J. Ferré, R. M. Marcé, F. Borrull and F. X. Rius, J. Chromatogr. A, 2004, 1035, 195–202 CrossRef CAS PubMed.
- K. István, R. Rajkó and G. Keresztury, J. Chromatogr. A, 2006, 1104, 154–163 CrossRef PubMed.
- J. M. Amigo, T. Skov, R. Bro, J. Coello and S. Maspoch, TrAC, Trends Anal. Chem., 2008, 27, 714–725 CrossRef CAS PubMed.
- B. Khakimov, J. M. Amigo, S. Bak and S. B. Engelsen, J. Chromatogr. A, 2012, 1266, 84–94 CrossRef CAS PubMed.
- F. Marini, A. D'Aloise, R. Bucci, F. Buiarelli, A. L. Magrì and A. D. Magrì, Chemom. Intell. Lab. Syst., 2011, 106, 142–149 CrossRef CAS PubMed.
- Y. L. Xie, J. J. Baeza-Baeza and G. Ramis-Ramos, Chemom. Intell. Lab. Syst., 1995, 27, 211–220 CrossRef CAS.
- J. M. Leitão and J. C. E. da Silva, Chemom. Intell. Lab. Syst., 2007, 89, 90–96 CrossRef PubMed.
- Z. Jiang, Z. Hao, Q. Wu, Y. Li, H. Liu and L. Yan, Drug Test. Anal., 2013, 5, 340–345 CrossRef CAS PubMed.
- P. Thongsrisomboon, B. Liawruangrath, S. Liawruangrath and S. Satienperakul, Food Chem., 2010, 123, 834–839 CrossRef CAS PubMed.
- P. Valderrama and R. J. Poppi, Anal. Chim. Acta, 2008, 623, 38–45 CrossRef CAS PubMed.
- R. Bro, PhD thesis, Københavns Universitet, 1998.
- A. Bozdoǧan, A. M. Acar and G. K. Kunt, Talanta, 1992, 39, 977–979 CrossRef.
- C. A. Andersson and R. Bro, Chemom. Intell. Lab. Syst., 2000, 52, 1–4 CrossRef CAS.
- R. P. H. Nikolajsen, K. S. Booksh, Å. M. Hansen and R. Bro, Anal. Chim. Acta, 2003, 475, 137–150 CrossRef CAS.
- S. Jawalekar, V. T. Survey and A. K. Bhutey, Int. J. Pharma Sci. Res., 2010, 1, 23–27 CAS.
- R. Yoshida and T. Ueki, NPG Asia Mater., 2014, 6, e107 CrossRef CAS.
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606–7616 CAS.
- M. Hara, C. C. Waraksa, J. T. Lean, B. A. Lewis and T. E. Mallouk, J. Phys. Chem. A, 2000, 104, 5275–5280 CrossRef CAS.
- W. L. Wallace and A. J. Bard, J. Phys. Chem., 1979, 83, 1350–1357 CrossRef CAS.
- E. Bolton and M. M. Richter, J. Chem. Educ., 2001, 78, 47 CrossRef CAS.
- B. Kansal and N. Singh, J. Indian Chem. Soc., 1978, 55, 304–307 CAS.
- R. Mehrotra and S. Ghosh, J. Chem. Phys., 1963, 224, 57–64 CAS.
- Z. He, R. Ma, Q. Luo, X. Yu and Y. Zeng, Anal. Chim. Acta, 1996, 54, 1003–1008 CAS.
| This journal is © The Royal Society of Chemistry 2015 |