
Synthesis of nanoporous hypercrosslinked polyaniline (HCPANI) for gas sorption and electrochemical supercapacitor applications†
Abstract
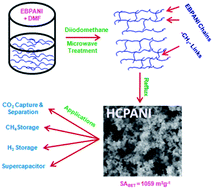
Nanoporous hypercrosslinked polyaniline with SABET of 1059 m2 g−1 has been synthesized. The specimen showed CO2 and CH4 uptake of 3.52 and 1.01 mmol g−1, respectively at 273 K, and H2 storage capacity of 1.85 wt% at 77 K and 1 bar. The material has a high specific capacitance of 410(±5) F g−1 and has shown good cyclability (100% retention) up to 1000 cycles.
 Please wait while we load your content...
Please wait while we load your content...

