
Metal-free iodine-mediated synthesis of vinyl sulfones at room temperature using water as solvent†
Ning Zhanga,
Daoshan Yang*a,
Wei Weia,
Li Yuana,
Youjuan Caob and
Hua Wang*a
aShandong Province Key Laboratory of Life-Organic Analysis, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, P. R. China. E-mail: yangdaoshan@tsinghua.org.cn; huawang_qfnu@126.com; Fax: +86-5374458306; Tel: +86-5374458306
bDepartment of Chemistry, Linyi Xijiao Experimental School, Linyi 276000, P. R. China
First published on 14th April 2015
Abstract
Metal-free efficient iodine-mediated synthesis of vinyl sulfones utilizing aryl sulfinates and alkenes has been realized under mild conditions in water. Notably, sodium methanesulfinate was used in this transformation, affording the β-iodo sulfones in good yields. This simple, efficient and environmentally benign transformation provides an attractive approach to various vinyl sulfones or β-iodo sulfones.
Introduction
Vinyl sulfone derivatives show a wide range of useful biological and pharmacological activities.1 As a consequence, development of more practical and efficient methods for the formation of these compounds is of great interest. Traditionally, Knoevenagel condensation of aromatic aldehydes with sulfonylacetic acids,2 β-elimination of selenosulfones or halosulfones,3 Wittig reaction of α-sulfinyl phosphonium ylides,4 and the oxidation of the corresponding sulfides5 are perhaps the most common. However, the preparation of vinyl sulfones with these methods depends on the availability of the requisite p-tolylsulfonylacetic acids, Wittig reagents or vinylic sulfides, and they are sometimes more difficult to prepare. Besides that, the multi-steps, strong bases and strong oxidants involving in these transformations might limit their wide applications. Transition-metal-catalyzed transformations are reliable tools in the synthetic chemistry.6 In recent years, Pd- or Cu-catalyzed cross-coupling of sulfinate salts with vinyl halides7 or alkenyl boronic acids8 leading to vinyl sulfones have made great progress. Very recently, Guo' group and Prabhu' group respectively developed a new copper-catalyzed method for the synthesis of vinyl sulfones through decarboxylative sulfonylation of cinnamic acids with sodium sulfinates (Scheme 1a).9a,b Despite some great advantages of these reactions, there are still certain limitations including harsh reaction conditions, the uneasily available precursors and especially toxic metal catalysts. As a result, there are increasing demands for metal-free reactions owing to trace-metal impurities can be avoided in the end products.10 For example, Jiang and co-workers developed an efficient metal-free approach for the synthesis of vinyl sulfones through cross-decarboxylative/coupling reaction of sodium sulfinates and cinnamic acids using dimethyl sulfoxide (DMSO) as solvent at 100 °C (Scheme 1b).11 | ||
| Scheme 1 Methods for the construction of vinyl sulfones. | ||
In view of the principles of green chemistry, the development of some synthesis methods under mild reaction conditions using environmental-friendly catalysts and medium has been being a challenging but attractive task in the current synthesis chemistry.12 As is well-known, replacement of common hazardous organic solvents by green and safe reaction media is an ongoing interest. In recent years, using water as an ideal medium in synthetic chemistry has attracted considerable attentions owing to its non-toxic characters. Importantly, water is also known to affect the selectivity and to enhance the reaction rates in organic transformations.13 As a wonderful example, Yadav and co-workers developed a LiBr catalysed one-pot method for synthesis of vinyl sulfones from terminal epoxides and sodium sulfinates in water (Scheme 1c).14 Gracefully excellent as this work could be, the terminal epoxides in some cases are not available, and the transformation is low regioselective. Alternatively, molecular iodine as an inexpensive, green and efficient reagent has been extensively used in organic transformation.15 In the present work, a metal-free molecular iodine-mediated synthesis approach has been developed for preparing vinyl sulfones from readily available alkenes and sodium sulfinates by using environmentally benign water as the solvent under mild conditions.
Results and discussion
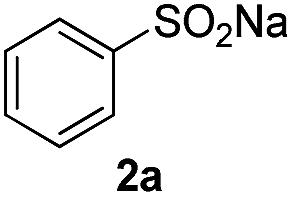
As shown in Table 1, phenylethylene (1a) and benzenesulfinic acid sodium salt (2a) were used as the model substrates to optimize the catalysis conditions including solvents and the amount of catalyst under air atmosphere. First, solvent (1 mL) including 1,2-dichloroethane, acetonitrile and methanol were investigated in the presence of 1 equiv. of iodine (relative to amount of 1a) at room temperature, and no obvious reaction activity was observed in this transformation, and we chose the most green and environmentally friendly water as the solvent. When the amount of the catalyst was changed to 0.5 equiv. from 2 equiv. the reaction yield decreased, only 46% yield was provided (Table 1, entry 4–7). No target product was observed in the absence of molecular iodine (Table 1, entry 8). After the optimization process of the amount of catalyst and solvent, the following reactions were carried out under our standard conditions: I2 (1.5 equiv.) and H2O (1 mL) under air atmosphere at room temperature for 2 h.
The scope of iodine-mediated reactions of the substituted alkenes with sodium sulfinates was investigated under the optimized conditions. As shown in Table 2, the tested substrates afforded good to excellent yields. For substituted alkenes, the substrates containing electron-withdrawing groups exhibited slightly higher reactivity than the others. Electron-effect of the substituted groups in sodium sulfinates including electron-rich, -neutral, -deficient groups did not display evidently difference of reactivity. The steric hindrance in alkenes such as 1-methyl-2-vinylbenzene, did not significantly affect the catalytic efficiency (Table 2, 3c and 3g). Interestingly, when sodium methanesulfinate was employed in this transformation, affording the β-iodo sulfones in good yields (Table 2, 3n and 3o), and no vinyl sulfones were observed. The reason might be the acidity of 2-H in the intermediate C is lower than aryl sodium sulfinates, which would not be easy to be attacked by iodine anion (see Scheme 4 formation mechanism). The cascade reactions could tolerate some functional groups such as methyl, C–Cl bond and C–Br bond, which could be used for further modifications at the substituted positions.
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 | 3 | Yield (%) |
| a Reaction conditions: the substituted alkenes (1 mmol), sodium sulfinates (1.5 mmol), molecular iodine (1.5 mmol), H2O (2 mL), at room temperature, 2 h, under air atmosphere.b Isolated yield. | ||||
| 1 |  |
 |
 |
94% |
| 2 |  |
2a | 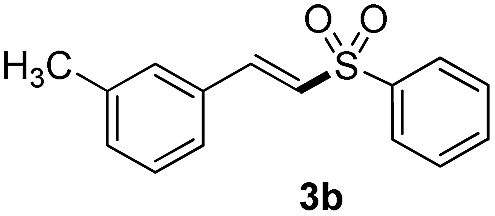 |
91% |
| 3 |  |
2a |  |
88% |
| 4 |  |
2a | 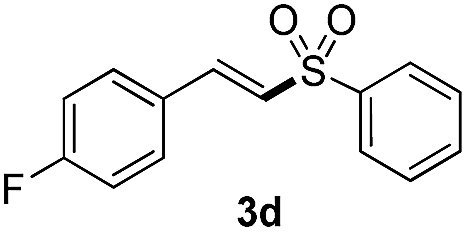 |
96% |
| 5 | 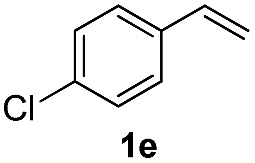 |
2a |  |
90% |
| 6 | 1a |  |
 |
90% |
| 7 | 1c | 2b |  |
85% |
| 8 | 1b | 2b |  |
87% |
| 9 | 1d | 2b |  |
91% |
| 10 | 1e | 2b |  |
84% |
| 11 | 1a |  |
 |
95% |
| 12 | 1b | 2c |  |
91% |
| 13 | 1e | 2c | 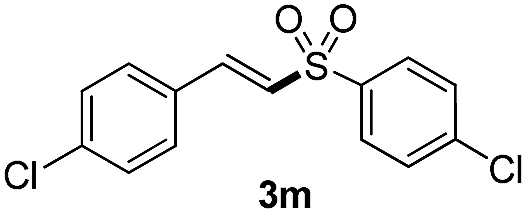 |
98% |
| 14 |  |
CH3SO2Na 2d |  |
93% |
| 15 | 1a | 2d | 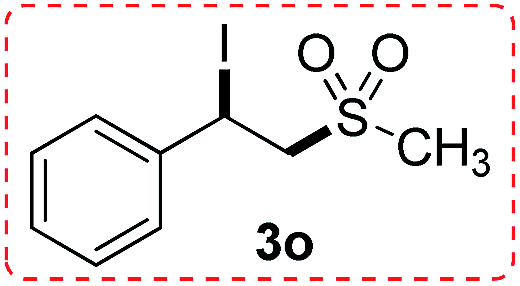 |
90% |

Further, we explored the synthetic applicability of the method. The gram-scale reaction was performed in the usual laboratory flask, and the reaction afforded 3j in 93% yield. As can be seen from Scheme 2, after two hours, the desired product 3j was completely precipitated in the flask owing to its poor solubility in water, and the pure 3j can be easily obtained through column chromatography after simple filtration. This example clearly demonstrates the practical aspect of this newly developed method.
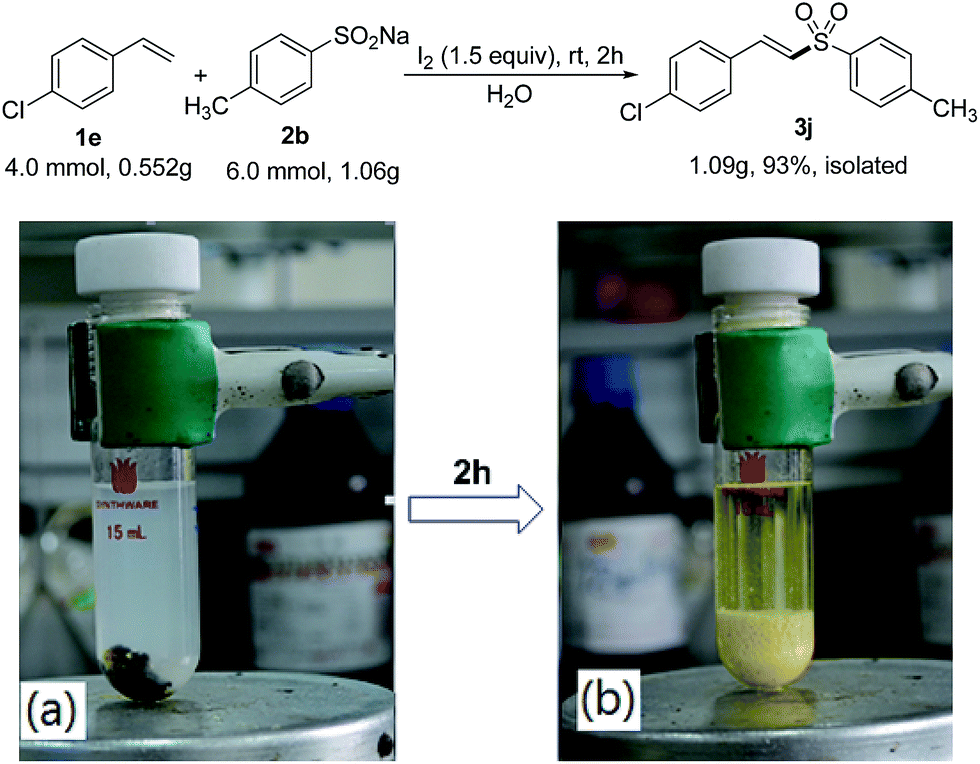 | ||
| Scheme 2 Synthesis of 3j on gram scale; reaction mixture (a) before and (b) after reaction. | ||
To understand the mechanism further, the reaction of 1a with 2a was carried out in the presence of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxy, a well-known radical inhibitor). As expected, the formation of 3a was completely inhibited in the reactions (Scheme 3), demonstrating that a radical process should be involved in this reaction.
 | ||
| Scheme 3 Control experiments. | ||
On the basis of these preliminary results above, together with literature reports,16 we proposed the mechanism in Scheme 4. Sodium sulfinate first interacted with molecular iodine to give sulfonyl iodide A, which then underwent homolytic cleavage to generate a sulfonyl radical. Addition of the sulfonyl radical to the substituted alkenes produced the reactive alkyl radical B, which could be trapped by iodine radical from homolytic cleavage the sulfonyl iodide or the molecular iodine to form intermediate C or the product 3n and 3o (if the R2 is alkyl). Elimination of HI from C finally generated the desired product 3a–3m (if the R2 is aryl).
 | ||
| Scheme 4 A proposed mechanism for the direct transformation. | ||
Experimental section
General
All reagents and solvents were obtained from commercial suppliers and used without further purification. Flash chromatography was performed on silica gel (200–300 mesh). 1H and 13C NMR data were recorded at 400 and 100 MHz on a BRUKER 400 spectrometer. Chemical shifts (δ) are expressed in parts per million (ppm) coupling constants (J) are in Hz. Proton and carbon magnetic resonance spectra (1H NMR and 13C NMR) were recorded using tetramethylsilane (TMS) in the solvent of CDCl3 as the internal standard (1H NMR: TMS at 0.00 ppm, CDCl3 at 7.28 ppm; 13C NMR: CDCl3 at 77.0 ppm).General procedure for synthesis of substituted vinyl sulfones
A 15 mL Schlenk tube equipped with a magnetic stirring bar was charged with I2 (1.5 equiv., 380 mg), alkenes (1.0 mmol), substituted various sodium sulfinates (1.5 mmol) and H2O (2 mL). The mixture was then stirred at room temperature for 2 h under air atmosphere, and then a saturated aqueous Na2S2O3 solution (2 mL) was added. Following that, the solution was extracted with ethyl acetate (15–20 mL). Finally, the combined organic phases were concentrated and the remaining residue was purified by column chromatography on silica gel to provide the desired product 3.(E)-(2-(Phenylsulfonyl)vinyl)benzene (3a)1
Eluent petroleum ether/ethyl acetate (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), pale yellow viscous liquid. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.97 (d, 2H, J = 8.0 Hz), 7.70 (d, 1H, J = 16 Hz), 7.63 (d, 1H, J = 8.0 Hz), 7.59–7.55 (m, 2H), 7.51–7.49 (m, 2H), 7.42–7.40 (m, 3H), 6.88 (d, 1H, J = 16 Hz). 13C NMR (CDCl3, 100 MHz, ppm) δ 142.5, 140.7, 133.4, 132.4, 131.3, 129.4, 129.1, 128.6, 127.7, 127.3. ESI-MS [M + H]+ m/z 245.2.
:1), pale yellow viscous liquid. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.97 (d, 2H, J = 8.0 Hz), 7.70 (d, 1H, J = 16 Hz), 7.63 (d, 1H, J = 8.0 Hz), 7.59–7.55 (m, 2H), 7.51–7.49 (m, 2H), 7.42–7.40 (m, 3H), 6.88 (d, 1H, J = 16 Hz). 13C NMR (CDCl3, 100 MHz, ppm) δ 142.5, 140.7, 133.4, 132.4, 131.3, 129.4, 129.1, 128.6, 127.7, 127.3. ESI-MS [M + H]+ m/z 245.2.
(E)-1-Methyl-3-(2-(phenylsulfonyl)vinyl)benzene (3b)2
Eluent petroleum ether/ethyl acetate (10:1), pale yellow solid, mp 104–106 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.96 (d, 2H, J = 8.0 Hz), 7.68 (d, 1H, J = 16 Hz), 7.63 (d, 1H, J = 8.0 Hz), 7.59–7.57 (m, 2H), 7.32–7.30 (m, 3H), 7.25–7.26 (m, 1H), 6.87 (d, 1H, J = 16 Hz), 2.37 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 142.7, 140.8, 138.9, 133.3, 132.4, 132.1, 129.3, 129.2, 129.0, 127.6, 127.0, 125.9, 21.3. ESI-MS [M + H]+ m/z 259.1.
(E)-1-Methyl-2-(2-(phenylsulfonyl)vinyl)benzene (3c)
Eluent petroleum ether/ethyl acetate (10:1), white solid, mp = 112–115 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.98–7.93 (m, 3H), 7.61–7.59 (m, 1H), 7.56–7.52 (m, 2H), 7.42 (d, 1H, J = 8.0 Hz), 7.27 (dd, 1H, J = 8.0 Hz), 7.21–7.27 (m, 2H), 6.78 (d, 1H, J = 16 Hz), 2.44 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 140.8, 140.2, 138.3, 133.4, 131.4, 131.1, 131.0, 129.4, 128.3, 127.7, 127.0, 126.6, 19.8. ESI-MS [M + H]+ m/z 259.1.
(E)-1-Fluoro-4-(2-(phenylsulfonyl)vinyl)benzene (3d)
Eluent petroleum ether/ethyl acetate (10:1). Pale yellow solid, mp = 110–112 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.97 (d, 2H, J = 8.0 Hz), 7.68 (d, 1H, J = 16 Hz), 7.63 (d, 1H, J = 8.0 Hz), 7.60–7.56 (m, 2H), 7.53–7.49 (m, 2H), 7.11 (t, 2H, J = 8.0 Hz), 6.81 (d, 1H, J = 16 Hz). 13C NMR (CDCl3, 100 MHz, ppm) δ 166.6, 163.1, 140.3 (d, J = 100.1 Hz), 133.5, 130.5 (d, J = 18.2 Hz), 129.4, 128.6 (d, J = 6.0 Hz), 127.7, 127.1 (d, J = 6.0 Hz), 116.5, 116.2. ESI-MS [M + H]+ m/z 263.2.
(E)-1-Chloro-4-(2-(phenylsulfonyl)vinyl)benzene (3e)3
Eluent petroleum ether/ethyl acetate (10:1). Colorless solid, mp = 127–129 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.97 (d, 2H, J = 8.0 Hz), 7.67 (d, 1H, J = 16 Hz), 7.60–7.56 (m, 3H), 7.44 (d, 2H, J = 8 Hz), 7.38 (d, 2H, J = 8 Hz), 6.87 (d, 1H, J = 16 Hz). 13C NMR (CDCl3, 100 MHz, ppm) δ 141.0, 140.7, 137.3, 133.5, 130.9, 129.8, 129.4, 128.9, 127.9, 127.7. ESI-MS [M + H]+ m/z 279.2.
(E)-1-Methyl-4-(styrylsulfonyl)benzene (3f)1
Eluent petroleum ether/ethyl acetate (10:1), pale yellow solid, mp 102–103 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.85 (d, 2H, J = 8.0 Hz), 7.68 (d, 1H, J = 16 Hz), 7.51–7.49 (m, 2H), 7.42–7.36 (m, 5H), 6.87 (d, 1H, J = 16 Hz), 2.46 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 144.4, 141.9, 137.8, 132.4, 131.1, 130.0, 129.1, 128.5, 127.7, 127.6, 21.6. ESI-MS [M + H]+ m/z 259.3.
(E)-1-Methyl-2-(2-tosylvinyl)benzene (3g)
Eluent petroleum ether/ethyl acetate (10:1), white solid, mp 126–127 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.94 (d, 1H, J = 16 Hz), 7.84 (d, 2H, J = 8 Hz), 7.65 (d, 1H, J = 16 Hz), 7.34 (d, 2H, J = 8 Hz), 7.32–7.29 (m, 3H), 6.85 (d, 1H, J = 16 Hz), 2.46 (s, 3H), 2.37 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 144.4, 139.7, 138.2, 137.9, 131.5, 131.1, 130.9, 130.1, 128.7, 127.8, 126.9, 126.5, 21.7, 19.9. ESI-MS [M + H]+ m/z 273.3.
(E)-1-Methyl-3-(2-tosylvinyl)benzene (3h)
Eluent petroleum ether/ethyl acetate (10:1), white solid, mp 82–83 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.93 (d, 1H, J = 16 Hz), 7.82 (d, 2H, J = 8 Hz), 7.41 (d, 1H, J = 8 Hz), 7.33 (d, 2H, J = 8 Hz), 7.25–7.21 (m, 1H), 7.20 (t, 2H, J = 8 Hz), 6.76 (d, 1H, J = 16 Hz), 2.44 (s, 3H), 2.43 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 144.3, 142.2, 138.8, 137.9, 132.4, 131.9, 129.9, 129.1, 128.9, 127.7, 127.4, 125.8, 21.6, 21.3. ESI-MS [M + H]+ m/z 273.3.
(E)-1-Fluoro-4-(2-tosylvinyl)benzene (3i)
Eluent petroleum ether/ethyl acetate (10:1), yellow oil. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.84 (d, 2H, J = 8 Hz), 7.63 (d, 1H, J = 16 Hz), 7.50–7.47 (m, 2H), 7.36 (d, 2H, J = 8 Hz), 7.11–7.06 (m, 2H), 6.81 (d, 1H, J = 16 Hz), 2.45 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 144.5, 140.7, 137.8, 130.7, 130.6, 130.1, 128.9, 127.8, 127.5, 116.5, 116.3, 21.7. ESI-MS [M + H]+ m/z 278.1.
(E)-1-Chloro-4-(2-tosylvinyl)benzene (3j)4
Eluent petroleum ether/ethyl acetate (10:1), white solid, mp = 140–141 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.84 (d, 2H, J = 8 Hz), 7.62 (d, 1H, J = 16 Hz), 7.43 (d, 2H, J = 8 Hz), 7.39–7.36 (m, 4H), 6.85 (d, 1H, J = 16 Hz), 2.46 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 144.6, 140.4, 137.5, 137.2, 131.0, 130.0, 129.7, 129.4, 128.3, 127.8, 21.6. ESI-MS [M + H]+ m/z 294.2.
(E)-1-Chloro-4-(styrylsulfonyl)benzene (3k)1
Eluent petroleum ether/ethyl acetate (10:1), pale yellow solid, mp = 85–87 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.91 (d, 2H, J = 8 Hz), 7.71 (d, 1H, J = 16 Hz), 7.56–7.52 (m, 4H), 7.45–7.42 (m, 3H), 6.86 (d, 1H, J = 16 Hz). 13C NMR (CDCl3, 100 MHz, ppm) δ 143.1, 140.1, 139.3, 132.2, 131.4, 129.7, 129.2, 128.7, 126.9, 125.7. HRMS m/z calcd for C14H12ClO2S [M + H]+: 279.0247, found: 279.0238.
(E)-1-(2-(4-Chlorophenylsulfonyl)vinyl)-3-methylbenzene (3l)
Eluent petroleum ether/ethyl acetate (10:1), colorless oil. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.86 (d, 2H, J = 4 Hz), 7.62 (d, 1H, J = 12 Hz), 7.50 (d, 2H, J = 4 Hz), 7.28–7.21 (m, 4H), 6.80 (d, 1H, J = 12 Hz), 2.35 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 143.4, 140.2, 139.4, 139.0, 132.3, 132.2, 129.7, 129.3, 129.2, 129.1, 126.7, 126.0, 21.3. ESI-MS [M + H]+ m/z 294.2.
(E)-1-Chloro-4-(2-(4-chlorophenylsulfonyl)vinyl)benzene (3m)
Eluent petroleum ether/ethyl acetate (10:1), pale yellow solid, mp = 164–166 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.90 (d, 2H, J = 8 Hz), 7.64 (d, 1H, J = 16 Hz), 7.56 (d, 2H, J = 8 Hz), 7.51–7.49 (m, 1H), 7.42–7.37 (m, 3H), 6.87 (d, 1H, J = 16 Hz). 13C NMR (CDCl3, 100 MHz, ppm) δ 141.5, 140.3, 139.0, 137.5, 130.6, 129.8, 129.7, 129.5, 129.2, 127.5. ESI-MS [M + H]+ m/z 313.1.
1-Chloro-3-(1-iodo-2-(methylsulfonyl)ethyl)benzene (3n)
Eluent petroleum ether/ethyl acetate (10:1), pale yellow solid, mp = 85–87 °C. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.52 (s, 1H), 7.42–7.39 (m, 1H), 7.33–7.31 (m, 2H), 5.53 (dd, 1H, J = 8 Hz), 4.14–4.08 (dd, 1H, J1 = 8 Hz, J2 = 12 Hz), 3.95–3.90 (dd, 1H, J1 = 8 Hz, J2 = 12 Hz), 2.51 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 143.0, 135.1, 130.6, 129.4, 127.7, 125.6, 65.4, 42.6, 15.5. ESI-MS [M + H]+ m/z 361.3.
(1-Iodo-2-(methylsulfonyl)ethyl)benzene (3o)
Eluent petroleum ether/ethyl acetate (10:1). Colorless oil. 1H NMR (CDCl3, 400 MHz, ppm) δ 7.53 (d, 1H, J = 8 Hz), 7.41–7.34 (m, 3H), 5.58 (dd, 1H, J = 4 Hz), 4.17 (dd, 1H, J1 = 8 Hz, J2 = 12 Hz), 3.94 (dd, 1H, J1 = 8 Hz, J2 = 4 Hz), 2.35 (s, 3H). 13C NMR (CDCl3, 100 MHz, ppm) δ 140.9, 129.4, 127.5, 125.7, 65.7, 42.4, 17.5. ESI-MS [M + H]+ m/z 311.1.
Conclusions
In conclusion, we have developed a highly efficient, green and practical iodine-mediated method for the synthesis of vinyl sulfones. The method enjoys the following advantages of: (a) commercially available and non-toxic molecular iodine as the catalyst; (b) environmentally friendly water as the solvent (c) no addition of any base or additive; (d) all the reactions performed at room temperature; (e) easy workup procedure; (f) outstanding tolerance of functional groups. All these merits can meet the requirements of green and sustainable chemistry. Therefore, the developed synthesis method will attract much attention in academic and industrial fields. Further investigation on the applications of this method is in progress.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (nos 21302110, 21375075 and 21302109), the Taishan Scholar Foundation of Shandong Province, the Project of Shandong Province Higher Educational Science and Technology Program (J13LD14), the Natural Science Foundation of Shandong Province (ZR2013BQ017), and the Scientific Research Foundation of Qufu Normal University (BSQD 2012021). We thank Xiao Zhu in this group for reproducing the results of 3a, 3k and 3r.References
- (a) G. Wang, U. Mahesh, G. Y. J. Chen and S. Yao, Org. Lett., 2003, 5, 737 CrossRef CAS PubMed; (b) M. Uttamchandani, K. Liu, R. C. Panicker and S. Q. Yao, Chem. Commun., 2007, 1518 RSC; (c) F. Hof, A. Schutz, C. Fah, S. Meyer, D. Bur, J. Liu, D. E. Goldberg and E. Diederich, Angew. Chem., Int. Ed., 2006, 45, 2138 CrossRef CAS PubMed; (d) D. C. Meadows, T. Sanchez, N. Neamati, T. W. North and J. Gervay-Hague, Bioorg. Med. Chem., 2007, 15, 1127 CrossRef CAS PubMed; (e) R. Ettari, E. Nizi, M. E. D. Francesco, M.-A. Dude, G. Pradel, R. Vicik, T. Schirmeister, N. Micale, S. Grasso and M. Zappala, J. Med. Chem., 2008, 51, 988 CrossRef CAS PubMed; (f) L. Mu, K. Drandarov, W. H. Bisson, A. Schibig, C. Wirz, P. A. Schubiger and G. Westera, Eur. J. Med. Chem., 2006, 41, 640 CrossRef CAS PubMed; (g) J. T. Palmer, D. Rasnick, J. L. Klaus and D. Brömme, J. Med. Chem., 1995, 38, 3193 CrossRef CAS PubMed.
- S. Chodroff and W. F. Whitmore, J. Am. Chem. Soc., 1950, 72, 1073 CrossRef CAS.
- (a) P. B. Hopkin and P. L. Fuchs, J. Org. Chem., 1978, 43, 1208 CrossRef; (b) R. A. Gancarz and J. L. Kice, Tetrahedron Lett., 1980, 21, 4155 CrossRef CAS.
- J. H. van Steenis, J. J. G. S. van Es and A. van der Gen, Eur. J. Org. Chem., 2000, 2787 CrossRef CAS.
- (a) A. A. Lindén, L. Krüger and J. Bäckvall, J. Org. Chem., 2003, 68, 5890 CrossRef PubMed; (b) Q. Xue, Z. Mao, Y. Shi, H. Mao, Y. Cheng and C. Zhu, Tetrahedron Lett., 2012, 53, 1851 CrossRef CAS.
- I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed.
- (a) W. Zhu and D. Ma, J. Org. Chem., 2005, 70, 2696 CrossRef CAS PubMed; (b) S. Cacchi, G. Fabrizi, A. Goggiamani, L. M. Parisi and R. Bernini, J. Org. Chem., 2004, 69, 5608 CrossRef CAS PubMed.
- F. Huang and R. A. Batey, Tetrahedron, 2007, 63, 7667 CrossRef CAS.
- (a) Q. Jiang, B. Xu, J. Jia, A. Zhao, Y.-R. Zhao, Y.-Y. Li, N.-N. He and C.-C. Guo, J. Org. Chem., 2014, 79, 7372 CrossRef CAS PubMed; (b) B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2014, 79, 8110 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Yuan, I. Thome, S. H. Kim, D. Chen, A. Beyer, J. Bonnamour, E. Zuidema, S. Chang and C. Bolm, Adv. Synth. Catal., 2010, 352, 2892 CrossRef CAS; (b) I. Thome, C. Besson, T. Kleine and C. Bolm, Angew. Chem., Int. Ed., 2013, 52, 7509–7513 CrossRef CAS PubMed; (c) G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 7195 CrossRef CAS PubMed.
- Y. Xu, X. Tang, W. Hu, W. Wu and H. Jiang, Green Chem., 2014, 16, 3720 RSC.
- (a) M. B. Gawande, P. S. Brancoa and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371 RSC; (b) R. B. Nasir Baig and R. S. Varma, Chem. Commun., 2013, 49, 752 RSC , and references therein.
- A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS PubMed.
- R. Chawla, R. Kapoor, A. K. Singh and L. D. S. Yadav, Green Chem., 2012, 14, 1308 RSC.
- (a) K. Zmitek, M. Zupan, S. Stavber and J. Iskra, Org. Lett., 2006, 8, 2491 CrossRef CAS PubMed; (b) T. Nobuta, N. Tada, A. Fujiya, A. Kariya, T. Miura and A. Itoh, Org. Lett., 2013, 15, 574 CrossRef CAS PubMed; (c) X. Pan, A. Boussonnière and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 14433 CrossRef CAS PubMed; (d) V. Humne, Y. Dangat, K. Vank and P. Lokhande, Org. Biomol. Chem., 2014, 12, 4832 RSC.
- P. Katrun, C. Mueangkaew, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, J. Org. Chem., 2014, 79, 1778 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5ra02927a |
| This journal is © The Royal Society of Chemistry 2015 |