
Silver nanoparticles: facile synthesis and their catalytic application for the degradation of dyes†
Kamaldeep Sharma‡
,
Gurpreet Singh‡,
Gurpreet Singh‡,
Manoj Kumar and
Vandana Bhalla*
Department of Chemistry, UGC Sponsored-Centre for Advanced Studies-I, Guru Nanak Dev University, Amritsar-143005, Punjab, India. E-mail: vanmanan@yahoo.co.in; Fax: +91 183 2258820; Tel: +91 183 2258802-9 ext. 3202
First published on 27th February 2015
Abstract
Derivatives 3, 5 and 7 having aldehyde groups at their periphery have been designed and synthesized. Interestingly, all three derivatives formed aggregates in aqueous media and served as reactors for facile synthesis of spherical silver nanoparticles (AgNPs) and silver nanorods at room temperature in aqueous media without the addition of any external reducing agents, stabilizing agents or shape directing additives. Interestingly, in situ generated AgNPs by aggregates of derivatives 3, 5 and 7 exhibit high catalytic degradation of industrially important organic dyes such as rhodamine B (RhB), methylene blue (MB), methyl orange (MO) and eriochrome black T (EBT).
1. Introduction
Aquatic systems are extremely important to human beings due to their utility as a source of water, in agricultural activities and for animal production. However, great damage to the aquatic environment is being caused due to direct discharge of various organic pollutants especially azo dyes which are widely used in the textiles, foodstuffs and paper industries.1,2 The uncontrolled discharge of azo dyes into the environment is alarming due to the toxic, mutagenic and carcinogenic characteristics of these dyes and their transformation products.3 These dyes can be removed by various processes such as flocculation,4 adsorption,5 ion exchange,6 UV radiation,7 ozonization,8 photochemical processes of electrochemical destruction9 etc., however, most of these methods suffer from high cost and lead to secondary pollution due to excessive use of chemicals. Therefore, development of simple and fast methods for the efficient degradation of organic dyes has become a challenging task for environmental chemists. Recently, noble metal NPs such as gold, silver and palladium have been used as catalyst for the degradation of various organic dyes.10–14 In comparison to gold and palladium, preparation of AgNPs is important due to its low cost, high photostability, abundance, environmentally benign nature and size and shape dependent catalytic properties.15–19 Among AgNPs of various shapes, preparation of quasi-spherical AgNPs has attracted lot of research interest due to their excellent catalytic efficiency. The literature reports show that utilization of additives such as polymers, oligoprolines, peptides and surfactants in the presence of reducing agent as the most popular approach for the generation of quasi spherical AgNPs.20–23 However, in most of these cases the additives act as strong complexing stabilizers which lead to generation of large and polydisperse NPs.24 Further, preparation of quasi-spherical AgNPs in aqueous media is difficult due to secondary nucleation arising from slow reduction of silver ions, hydrolysis of silver ions and decomposition of newly formed AgNPs.20 Thus, development of simple and fast method for generation of stable quasi-spherical monodisperse AgNPs in aqueous media remains a challenge.Our research work involves development of supramolecular aggregates in aqueous media and their utilization as reactors for the preparation of metal NPs such as gold, palladium and iron.25–28 Recently, we reported aggregates of pentacenequinone derivatives having pyridine/nitrile/amino groups which served as reactors for the preparation of palladium and gold NPs.25–27 Considering the high price of palladium and gold, silver is unique due to its excellent qualities in terms of plasmonic ability, available nanostructures and material cost.17 Thus, keeping in view the advantages of AgNPs, in the present manuscript, we have designed and synthesized pentacenequinone derivative 3 having aldehyde groups. Pentacenequinone is the scaffold of our choice as it can be functionalized with different moieties at the periphery and in an aqueous environment pentacenequinone derivatives are known to form aggregates which can serve as reactors and stabilizers for the preparation of metal NPs.25–27 For generation of AgNPs, we planned to reduce the Ag+ ions with the help of aldehyde groups that are present at the periphery of the molecules. We envisioned that during the reduction process, aldehyde groups will be oxidized to carboxylic acids and the in situ generated AgNPs will be stabilized due to their interactions with the oxidized form of aggregates of derivative 3.29,30 To study the influence of scaffold on size/shape and catalytic efficiency of in situ generated AgNPs, we also synthesized hexaphenylbenzene (HPB) and perylene diimide (PDI) based derivatives 5 and 7 having aldehyde groups at their periphery. Being electron rich, HPB derivative having high reduction potential31 (vide infra) is expected to influence rate of formation of AgNPs. On the other hand, PDI derivatives are known for their electron acceptor nature and low reduction potential.32 Thus, we were interested to know whether the aggregates of PDI derivative 7 will be successful in generating AgNPs or not. Interestingly, all the derivatives 3, 5 and 7 formed fluorescent aggregates in aqueous media and these aggregates served as reactors for the generation of AgNPs. Furthermore, the in situ generated NPs exhibited good catalytic efficiency for the degradation of industrially important organic dyes such as RhB, MO, EBT and MB. Thus, the present study has several advantages. First, supramolecular aggregates of electron rich/electron deficient molecules have been used at room temperature for generation of AgNPs without the addition of any external reducing agents, stabilizing agents or shape directing additives. Second, effect of scaffold and morphology/nature of aggregates on rate of generation of AgNPs has been studied and it is found that despite the low reduction potential of aggregates of PDI derivative 7 exhibit high rate of formation of AgNPs due to its porous morphology. Third, the present study demonstrates the catalytic efficiency of aggregates of pentacenequinone/HPB/PDI stabilized AgNPs for degradation of organic dyes.
2. Results and discussion
Derivative 3 was synthesized by Suzuki–Miyaura coupling of 2,3-dibromopentacenequinone 1,33 with 4-formylphenylboronic acid 2 (Scheme 1). The structure of derivative 3 was confirmed from its spectroscopic and analytical data (Fig. S37 and S38 in ESI†). The 1H NMR spectrum of derivative 3 showed four singlets at 10.03, 9.04, 8.99 and 8.23 ppm, corresponding to the aldehyde group and aromatic protons, respectively, two doublets at 7.83 and 7.43 ppm and two multiplets at 7.74–7.75 and at 8.15–8.17 ppm, corresponding to aromatic protons (Fig. S37 in ESI†). The mass spectrum of derivative 3 showed a parent ion peak at m/z 517.1305 (M + 1)+ (Fig. S38 in ESI†). These spectroscopic data corroborate the structure 3 for this compound.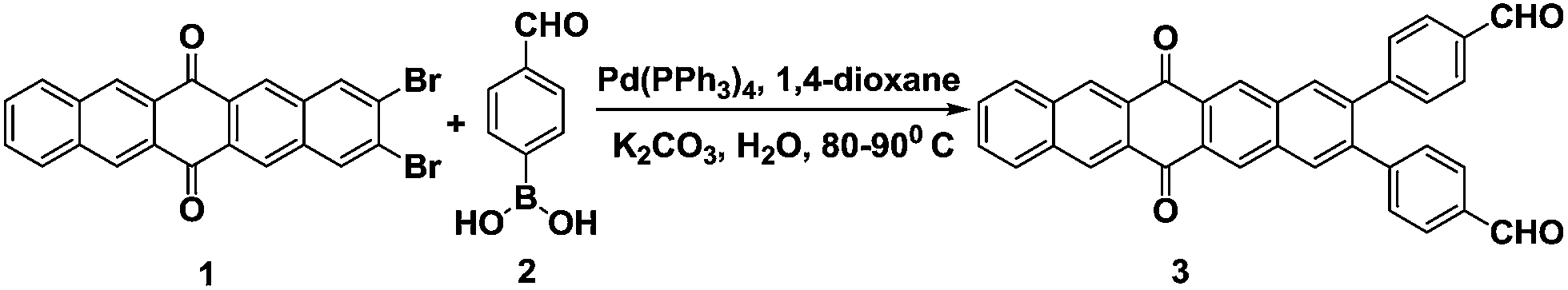 | ||
| Scheme 1 Synthesis of pentacenequinone based derivative 3. | ||
The UV-vis spectrum of derivative 3 in THF exhibits absorption bands at 315 and 406 nm due to π–π* and n–π* transitions. On addition of water (≤70% volume fractions) to the THF solution of derivative 3, a new absorption band is observed at 422 nm (Fig. 1A). This new band may be attributed to the formation of intermolecular charge transfer (ICT) state as the molecules of derivative 3 are packed more closely during the aggregation process.34,35 Further increase of water fraction up to 95% leads to the appearance of level-off tail (inset Fig. 1A). The presence of the level-off tail in the visible region suggests the formation of aggregates. The scanning electron microscopy (SEM) image of derivative 3 in the solvent mixture of H2O/THF (9.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5) showed flake like morphology of aggregates (Fig. 1B).
:0.5) showed flake like morphology of aggregates (Fig. 1B).
 | ||
| Fig. 1 (A) UV-vis spectra showing the variation of absorbance of derivative 3 (10 μM) in H2O/THF mixture with different fractions of H2O. Inset: enlarge UV-vis spectra of derivative 3 (10 μM) with the addition of H2O/THF mixture in the range of 425–600 nm showing level-off tail (B) SEM image of derivative 3 in H2O/THF (9.5/0.5) mixture showing flake like morphology; scale bar 2 μm. | ||
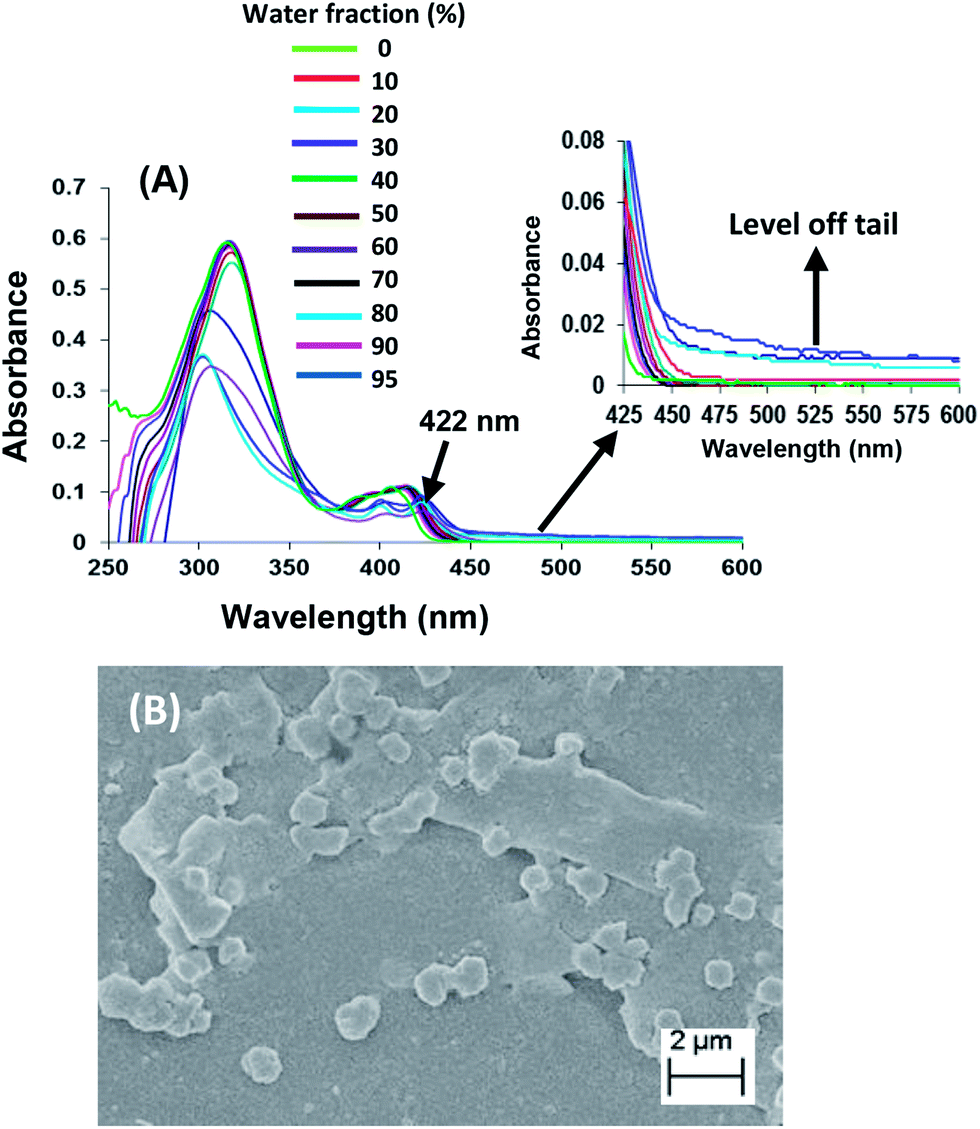
The recognition behaviour of aggregates of derivative 3 (10 μM) in H2O/THF (9.5/0.5) was studied toward different metal ions such as Cd2+, Hg2+, Fe2+, Ni2+, Zn2+, Cu2+, Pb2+, Ca2+, Co2+, Au3+, Na+, K+, Li+, Pd2+, Cr3+, Al3+ ions as their perchlorate/chloride or both perchlorate and chloride salts by UV-vis spectroscopy (Fig. S1 and S2 in ESI†). Upon addition of Ag+ ions (100 equiv.) to the solution of derivative 3 in mixed aqueous media (H2O/THF, 9.5/0.5), a broad absorption band at 470 nm was observed after 4 hours (Fig. 2) which is the characteristic surface plasmon resonance (SPR) of spherical AgNPs.36 In comparison to the reported absorption band of AgNPs (ca. 390 nm), the SPR band is red shifted.37 Further, instead of well-known sharp peak for AgNPs, a broad peak is observed. The presence of broad and red shifted band may be attributed to the interactions between AgNPs and aggregates of derivative 3.38 The intensity of this band increases with time and the rate constant39 for the formation of AgNPs was found to be 8.31 × 10−5 s−1 (Page S6 in ESI†).40,41
 | ||
| Fig. 2 UV-vis spectra of derivative 3 (10 μm) upon addition of Ag+ ions (100 equiv.) at different time intervals in H2O/THF (9.5/0.5). Inset: photograph of solution of AgNPs recorded after 4 hours. | ||
The formation of AgNPs is also supported by powder X-ray diffraction (XRD) analysis of the sample containing AgNPs of derivative 3 (Fig. S4 in ESI†) which shows peaks corresponding to diffraction from the 111, 200, 220, 311 crystal planes of fcc silver(0).42
To get insight into the mechanism of formation of AgNPs, we slowly evaporated the solution of aggregates of derivative 3 containing AgNPs. After several days, precipitates were observed which were filtered and washed with THF. The 1H NMR spectrum of the residue obtained after evaporation of THF was recorded and it showed the absence of signal corresponding to aldehyde protons in derivative 3 with slight upfield shift of aromatic signals (Fig. S5 in ESI†). Further, FTIR spectrum of the resulting residue showed the presence of a new band at 1760 cm−1 which may be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of acid (Fig. S6 in ESI†). These results suggest the involvement of aldehyde groups in the process and their subsequent oxidation to acid.29 The chemical composition of the resulting precipitates was determined by energy dispersive X-ray (EDX) analysis. The presence of a peak of Ag in EDX spectrum of resulting precipitates indicates the presence of Ag metal in the precipitates40 (Fig. S7 in ESI†). We also studied the behaviour of derivative 3 towards Ag+ ions in THF but absorption spectra did not indicate any absorption band corresponding to AgNPs (Fig. S8 in ESI†). These studies suggest the importance of water in the preparation of AgNPs. To evaluate the role of pentacenequinone moiety for the formation of AgNPs, we tested the generation of AgNPs using benzaldehyde. Upon addition of Ag+ ions (100 equiv.) to the solution of benzaldehyde in mixed aqueous media (H2O/THF, 9.5/0.5), no absorption band corresponding to AgNPs was observed in the visible region (Fig. S9 in ESI†). This study highlights the importance of aggregates of pentacenequinone based derivative 3 for the preparation of AgNPs. On the basis of above results, we suggest that upon addition of Ag+ ions to the solution of aggregates of derivative 3, the silver ions enter the network of interconnected channels, interact with aldehyde moieties and get reduced. Thus, aggregates of derivative 3 function as reducing agents as well as stabilizing agents for the preparation of AgNPs.43
O group of acid (Fig. S6 in ESI†). These results suggest the involvement of aldehyde groups in the process and their subsequent oxidation to acid.29 The chemical composition of the resulting precipitates was determined by energy dispersive X-ray (EDX) analysis. The presence of a peak of Ag in EDX spectrum of resulting precipitates indicates the presence of Ag metal in the precipitates40 (Fig. S7 in ESI†). We also studied the behaviour of derivative 3 towards Ag+ ions in THF but absorption spectra did not indicate any absorption band corresponding to AgNPs (Fig. S8 in ESI†). These studies suggest the importance of water in the preparation of AgNPs. To evaluate the role of pentacenequinone moiety for the formation of AgNPs, we tested the generation of AgNPs using benzaldehyde. Upon addition of Ag+ ions (100 equiv.) to the solution of benzaldehyde in mixed aqueous media (H2O/THF, 9.5/0.5), no absorption band corresponding to AgNPs was observed in the visible region (Fig. S9 in ESI†). This study highlights the importance of aggregates of pentacenequinone based derivative 3 for the preparation of AgNPs. On the basis of above results, we suggest that upon addition of Ag+ ions to the solution of aggregates of derivative 3, the silver ions enter the network of interconnected channels, interact with aldehyde moieties and get reduced. Thus, aggregates of derivative 3 function as reducing agents as well as stabilizing agents for the preparation of AgNPs.43
To get insight into effect of morphology and nature of aggregates on the rate of formation, size and shape of AgNPs, we also designed and synthesized HPB and PDI based derivatives 5 and 7 having aldehyde groups at the periphery. Derivatives 5 and 7 were synthesized by Suzuki–Miyaura cross-coupling of HPB based derivative 444 and PDI based derivative 645 with 4-formyl phenyl boronic acid 2 in 70% and 50% yields, respectively (Scheme 2).
 | ||
| Scheme 2 Synthesis of HPB and PDI based derivatives 5 and 7. | ||
The structures of derivatives 5 and 7 were confirmed from their spectroscopic and analytical data (Pages S36–S41 in ESI†). The UV-vis studies of derivative 5 and 7 in H2O/THF mixture show the presence of aggregates (Fig. S10 and S11 in ESI†). The SEM images of the derivatives 5 and 7 in H2O/THF solution show the presence of disc shaped and irregular shaped aggregates with porous morphology, respectively (Fig. S12 and S13 in ESI†). The porous morphology of derivative 7 was confirmed from transmission electron microscopic (TEM) images46 (Fig. S14 in ESI†). The absorption studies of derivatives 5 and 7 (10 μM) in H2O/THF solution were carried out towards Ag+ ions and it was found that aggregates of both derivatives served as nanoreactors for preparation of AgNPs (Fig. S15 and S16 in ESI†). In both cases, plasmon absorption is observed in the region 430–460 nm which indicates the formation of AgNPs36 which was supported by their powder XRD analysis. (Fig. S17 and S18 in ESI†). We also investigated the electrochemical behavior of the derivatives 3, 5 and 7 in the presence of AgNO3 in H2O/ACN (1/9, v/v). The cyclic voltammetric (CV) measurements of derivative 3/5/7 in the presence of 0.1 M tetrabutylammonium perchlorate as supporting electrolyte (Page S18 in ESI†), exhibit cathodic peak which corresponds to the reduction of the silver ions Ag(I) to Ag(0).47 Interestingly, the voltammogram of derivatives 3 and 5 in the presence of silver ions show the trace crossing, thus, suggesting that rate determining step for reduction of Ag+ ions on electrode is nucleation.48 No trace crossing was observed in the case of CV of derivative 7 in presence of silver ions. On the basis of these CV studies, derivative 5 was found to have high reduction potential (Table S3, in ESI†), thus, it is expected to show higher rate constant for the formation of AgNPs. However, on the basis of absorption studies, the rate constants for the formation of AgNPs in case of aggregates of derivatives 3, 5 and 7 were found to be 8.3 × 10−5 s−1, 4.56 × 10−5 s−1 and 7.1 × 10−5 s−1, respectively. These results show that among the derivatives 3/5/7 aggregates of derivative 3 display higher rate constant for the formation of AgNPs. We believe that flake like morphology of aggregates of derivative 3 provide large surface area of contact which results in stronger interactions with metal ions, hence, faster rate of reduction is observed in this case. Further, despite the presence of electron deficient scaffold, aggregates of derivative 7 exhibit high rate constant for the formation of AgNPs. We believe that due to the porous morphology and presence of greater number of heteroatoms in the molecule, aggregates of derivative 7 bind strongly with silver ions and form a uniform layer around them; hence, rate of reduction is faster.49 These results suggest that morphology as well as reduction potential of the reducing/stabilizing agent greatly influences the rate of formation of AgNPs.
The TEM images of AgNPs generated by aggregates of derivatives 3 and 7 showed the presence of spherical AgNPs (Fig. 3A and B) whereas TEM image of AgNPs generated by aggregates of derivative 5 showed the presence of nanospheres and nanorods (Fig. 3C and D).
 | ||
| Fig. 3 (A and B) TEM images of spherical AgNPs of derivative 3 and 7 (C) AgNPs and (D) nanorods generated by aggregates of derivative 5. Inset: their corresponding electron diffraction patterns of a nanosized region containing AgNPs of derivative 3, 5, 7; scale bar (A) 20 nm (B) 50 nm (C) 100 nm and (D) 50 nm. | ||
We believe that due to higher rate of formation of AgNPs in case of aggregates of derivatives 3 and 7, higher numbers of reduced AgNPs are available which lead to uniform growth of nanoparticles in all directions, thus, spherical nanoparticles are obtained.50
Earlier, KI has been used to tailor the growth of AgNPs into a quasi-spherical shape via its preferential adsorption on the (111) facets of nanorods.51 Thus, we studied the effect of iodide on the shape of AgNPs in case of aggregates of derivatives 3, 5 and 7. The UV-vis spectrum of AgNPs generated by aggregates of derivative 5 in the presence of KI exhibited more symmetric SPR band in comparison to that observed in absence of KI (Fig. 4A). The observed rate constant for the formation of AgNPs by derivative 5 in the presence of KI was found to be 1.4 × 10−4 s−1 (Fig. S20 in ESI†). The TEM image of AgNPs generated by aggregates of derivative 5 in the presence of KI, showed the presence of only spherical AgNPs and no nanorods were observed (Fig. 4B). These results highlight the role of iodide ions in tailoring of growth of AgNPs.
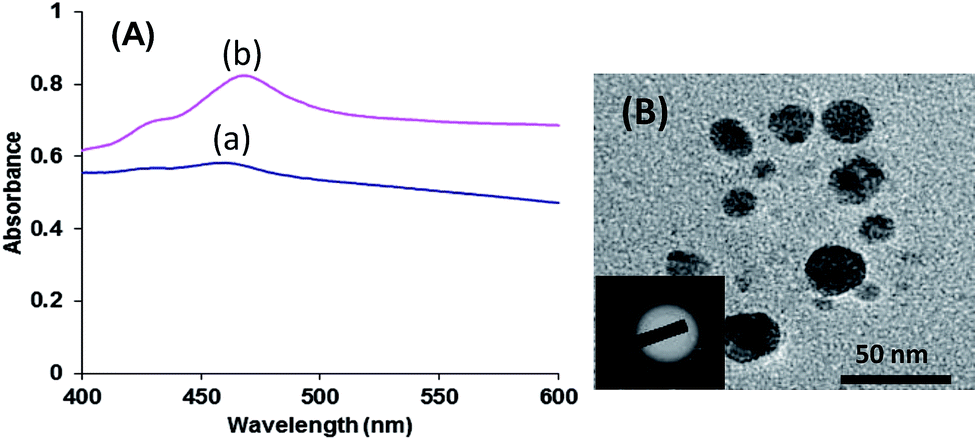 | ||
| Fig. 4 (A) UV-vis spectra of AgNPs of derivative 5 (10 μm) (a) before and (b) after addition of KI. (B) TEM image quasi-spherical AgNPs of derivative 5 after addition of KI. Inset: electron diffraction pattern of a nanosized region containing solution of AgNPs of derivative 5; scale bar 50 nm. | ||
Further, we investigated the catalytic activities of in situ generated AgNPs of aggregates of derivatives 3, 5 and 7 in the electron-transfer reaction between organic dyes viz. RhB, MB, MO, EBT and borohydride ions. For each reaction, three different samples were studied i.e. in situ generated AgNPs by aggregates of 3, in situ generated AgNPs by aggregates of 5 in presence of iodide ions and in situ generated AgNPs by aggregates of 7. The progress of the each catalytic dye degradation was monitored through changes in the UV-vis spectra with time as reduction proceeded (Fig. S21–S31 in ESI†). All the reactions offered a good correlation with first order kinetics.52 First order rate constants were determined from linear regressions of relative absorbance of dyes plotted versus time (Fig. S21–S31 in the ESI†).
The catalytic degradation of RhB in the presence of in situ generated AgNPs by aggregates of derivative 3 with time is presented in Fig. 5. The catalytic degradation process was monitored through the change in intensity of the characteristic absorption peak of RhB at 554 nm. The absorption spectrum of RhB solution showed time-dependent change in the presence of AgNPs under visible-light irradiation. The catalytic degradation process was monitored through the change in intensity of the characteristic absorption peak of RhB at 554 nm. The characteristic absorption peak of RhB dye at 554 nm rapidly decreased in intensity as time prolonged and it disappeared completely after about 2.5 min (Fig. 5) and the colour of the solution changed from the initial pink-red to almost colourless. The in situ generated AgNPs by aggregates of derivatives 3 and 5 were found to exhibit higher catalytic efficiency towards degradation reaction of RhB as compared to AgNPs of derivative 7 (Table 1). Overall, the degradation rate constant of aggregates of derivatives 3 stabilized AgNPs for the degradation of RhB dye was found to be better than the rate constants reported in literature.53
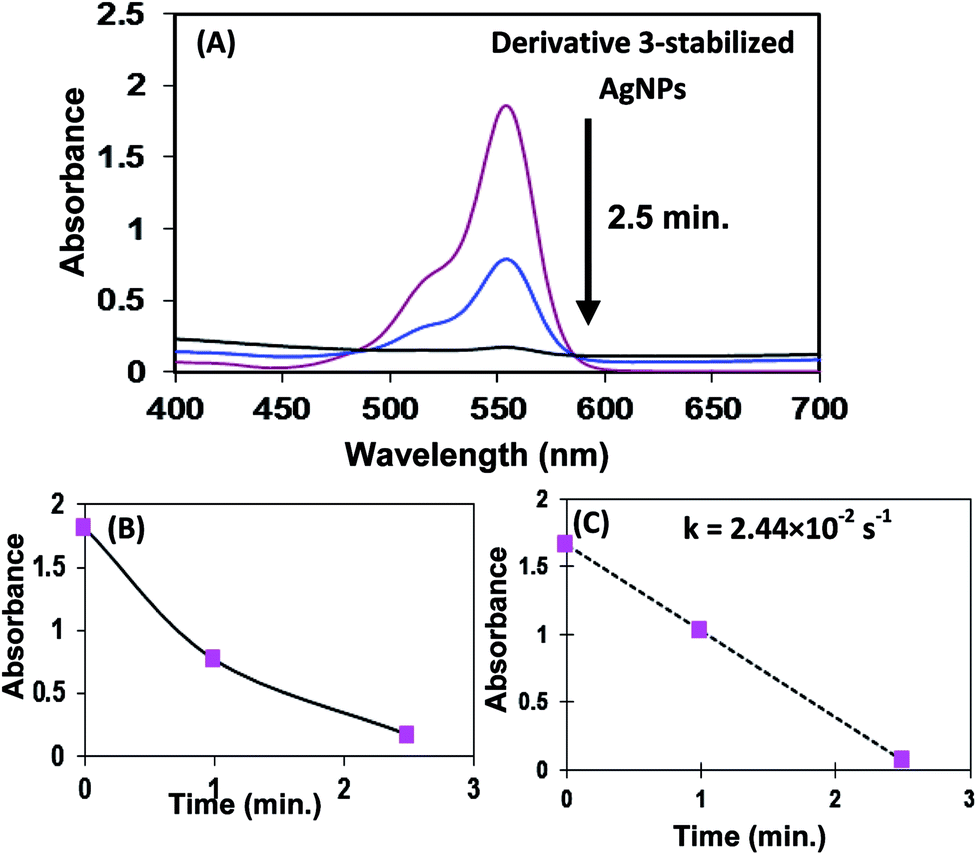 | ||
| Fig. 5 Catalytic properties of in situ generated AgNPs by aggregates of 3. (A) Decrease in rhodamine B absorbance at 554 nm over time using derivative 3-stabilized AgNPs as catalyst in the presence of NaBH4 (B) normalized kinetic trace of the absorbance at 554 nm during the reduction of rhodamine B. (C) Kinetic rate constant. | ||
| Dyes | RhB | MO | MB | EBT |
|---|---|---|---|---|
| Rate constants for degradation of dyes by in situ generated AgNPs of derivative 3 | 2.44 × 10−2 s−1 | 3.05 × 10−3 s−1 | 2.81 × 10−3 s−1 | 3.2 × 10−3 s−1 |
| Rate constants for degradation of dyes by in situ generated AgNPs of derivative 5 in the presence of KI | 1.8 × 10−2 s−1 | 5.9 × 10−3 s−1 | 3.8 × 10−3 s−1 | 4.3 × 10−3 s−1 |
| Rate constants for degradation of dyes by in situ generated AgNPs of derivative 7 | 6.6 × 10−3 s−1 | 5 × 10−3 s−1 | 2.4 × 10−3 s−1 | 1.88 × 10−3 s−1 |
The enhanced degradation observed in case of these nanoparticles may be attributed to several reasons, first, spherical shape and small size of generated nanoparticles and secondly, the aggregates of derivatives 3/5/7 having heteroatoms assist in bringing the dye molecules closer to catalytic sites, thus resulting in faster degradation process.
The photoefficiency of aggregates of derivative 3 stabilized AgNPs was also checked for photodegradation of RhB under illumination of visible light in the absence of borohydride ions (Fig. 6).
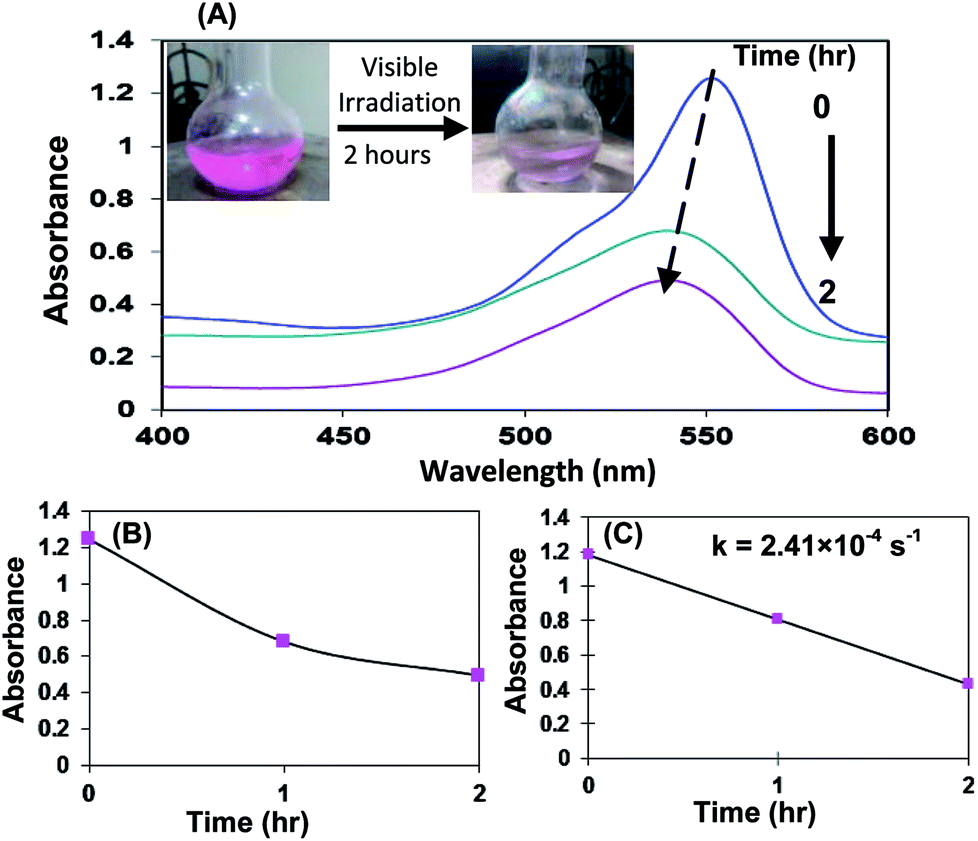 | ||
| Fig. 6 (A) UV-vis spectral changes of RhB over time using derivative 3-stabilized AgNPs as catalyst under visible light irradiations. Inset: photographs show colour change of aqueous RhB solution after 2 hours under visible light irradiations (B) normalized kinetic trace of the absorbance at 554 nm during the photodegradation of RhB. (C) Kinetic rate constant. | ||
Visible light irradiation of the aqueous RhB/AgNPs solution leads to a decrease in absorption with a wavelength shift of the band to shorter wavelengths. The degradation ratio was calculated using the following equation:54
| D(%) = (C0 − C)/C0 × 100% = (A0 − A)/A0 × 100% |
To get insight in to the photocatalytic mechanism, we carried out RhB photodegradation with AgNPs in the presence of 10% of triethanolamine (TEOA). The photodegradation rate was dramatically decreased in the presence of TEOA and after 2 h only 38% degradation was observed with no shift in the position of absorption peak corresponding to RhB (Fig. S32 in ESI†). This result suggests that the photodegradation mechanism involves hole transfer from the photoactive material to the solution.57
To test the reusability of these nanoparticles, the catalytic degradation of RhB in the presence of NaBH4 aqueous solution was chosen as a model reaction. After the catalytic degradation of RhB, colour changed from pink to colourless. The reaction mixture containing AgNPs catalyst was subjected to next catalytic sequence by adding aqueous solution of RhB and NaBH4 to reaction mixture. The complete degradation of RhB occurs up to three cycles in the presence of AgNPs of aggregates of derivative 3 (Fig. S33 in ESI†). On the other hand, aggregates of derivatives 5 and 7 stabilized AgNPs show complete degradation of RhB upto three and five cycles, respectively (Fig. S34 and S35 in ESI†). Further, recyclability of AgNPs generated by aggregates of derivative 3 as heterogeneous catalyst was checked by using the same reaction as model reaction. For this the AgNPs were precipitated after the completion of reaction by adding dichloromethane to the reaction mixture. The precipitated AgNPs were filtered and reused at least three times using this recycling procedure for identical reaction.
Other non-biodegradable dyes, such as MB, MO and EBT were also examined using AgNPs of aggregates of derivatives 3, 5 and 7 as catalysts (Table 1). In the presence of aggregates of 3 stabilized AgNPs, the colouration of methylene blue is visibly reduced within 18 minutes (Fig. S21 in ESI†). The intensity of characteristic wavelength of MB at 665 nm is gradually decreased as the reaction proceeds and nearly 100% reduction is observed. Under similar conditions as used for degradation of RhB, we also carried out degradation of MO and EBT using in situ generated AgNPs of aggregates of derivatives 3, 5, 7 and results are summarised in Table 1.
A blank experiment was carried out for the degradation of RhB, MB, MO and EBT with NaBH4 in the absence of AgNPs and no change in absorption corresponding to each dye was observed after 24 hours (Fig. S36 in ESI†).
3. Experimental section
3.1. Instruments
UV-vis spectra were recorded on a SHIMADZU UV-2450 spectrophotometer, with a quartz cuvette (path length, 1 cm). The cell holder was thermostatted at 25 °C. The fluorescence spectra were recorded on a SHIMADZU 5301 PC spectrofluorimeter. Scanning electron microscope (SEM) images were obtained with a field-emission scanning electron microscope (SEM CARL ZEISS SUPRA 55). TEM images were recorded from Transmission Electron Microscope (TEM) – JEOL 2100F. Elemental analysis was performed on a Flash EA 1112 CHNS/O analyzer of Thermo Electron Corporation. The 1H and 13C NMR spectra were recorded on a JEOL-FT NMR-AL 300 MHz and Bruker-FT NMR-AL 500 MHz spectrophotometers using CDCl3 and DMSO-d6 as solvent and tetramethylsilane (SiMe4) as internal standards. UV-vis studies were performed in THF and H2O/THF mixture. Data are reported as follows: chemical shifts in ppm (δ), multiplicity (s = singlet, d = doublet, br = broad singlet, m = multiplet), coupling constants J (Hz), integration and interpretation.3.2. Materials
All reagents were purchased from Aldrich and were used without further purification. THF was dried over sodium and benzophenone and kept over molecular sieves overnight before use. Silica gel (60–120 mesh) was used for column chromatography.3.3. Synthesis
:30 (chloroform:hexane) as an eluent to give 0.191 g (35%) of compound 3 as yellow solid; m.p. > 260 °C; 1H NMR (500 MHz, CDCl3): δ = 7.42 [d, 4H, J = 5.0 Hz, ArH], 7.74–7.76 [m, 2H, ArH], 7.83 [d, 4H, J = 10.0 ArH], 8.15–8.17 [m, 2H, ArH], 8.23 [s, 2H, ArH], 8.99 [s, 2H, ArH]; 9.04 [s, 2H, ArH]; 10.03 [s, 2H, CHO]; TOF MS ES+: 517.1305 [M + 1]+; Elemental analysis: calcd for C36H20O4: C 83.71; H 3.55; O 12.39 found: C 83.68; H 3.51; O 12.35. Due to poor solubility of compound 3, its 13C NMR spectrum could not be recorded.:90) (hexane/chloroform) as an eluent to give 0.255 g (60%) of compound 5, recrystallized from methanol as pale yellow solid; mp: 270–272 °C; 1H NMR (300 MHz, CDCl3, ppm). δ = 9.97 (S, 2H, CHO), 7.82 (d, J = 9 Hz, 4H, Ar-H), 7.57 (d, J = 9 Hz, 4H, Ar-H), 7.19 (d, J = 9 Hz, 4H, Ar-H), 6.98 (d, J = 6 Hz, 4H, Ar-H), 6.94–6.87 (m, 20H, Ar-H); 13C NMR (CDCl3, 75 MHz, ppm) 192.53, 147.12, 141.64, 141.30, 140.96, 140.85, 139.95, 136.55, 135.32, 132.58, 131.83, 130.55, 127.65, 127.25, 127.13, 126.05, 125.89, 125.80; ESI/MS, m/z = 765.2743 [M + Na]+. Elemental analysis: calculated for C56H38O2: C 90.54; H 5.16; O 4.31; found: C 90.47%; H 5.12%; N 4.29%.:90) (hexane/chloroform) as an eluent to give 0.265 g (50%) of compound 7, recrystallized from methanol as dark red solid; mp: 265–267 °C. 1H NMR (300 MHz, CDCl3, ppm). δ = 10.15 (s, 2H, CHO), 8.61 (s, 2H, Ar-H), 8.18 (d, J = 9 Hz, 2H, Ar-H), 8.05 (d, J = 9 Hz, 4H, Ar-H), 7.76 (d, J = 9 Hz, 4H, Ar-H), 7.65 (d, J = 9 Hz, 2H, Ar-H), 4.18 (t, J = 6 Hz, 4H), 1.72 (br, 4H), 1.25 (br, 30H), 0.87 (t, J = 6 Hz, 12H); 13C NMR (CDCl3, 125 MHz, ppm) 191.48, 163.14, 148.00, 139.66, 136.14, 134.85, 132.48, 131.51, 130.75, 130.36, 127.87, 129.95, 122.62, 122.34, 117.65, 40.65, 31.80, 29.52, 29.26, 28.00, 26.98, 22.57, 14.06; Maldi TOF MS ES+: 936.5 [M + 2]+. Elemental analysis: calculated for C62H66N2O6: C 79.63; H 7.11; N 3.00; O 10.26; found: C 79.60%; H 7.09%; N 2.96%; O 10.21%.3.4. Catalytic degradation of rhodamine B
The catalytic degradation of RhB was carried out at room temperature in the presence of aqueous solution of AgNPs of derivatives 3, 5 and 7. Firstly, same volume of aqueous solution of NaBH4 (0.2 M) and RhB (2 mM) solution were mixed to obtain the mixture solution of NaBH4 and RhB dye and diluted 100 times in deionized water. Addition of catalytic amount of AgNPs solution generated by aggregates of derivative 3 (3 μL, 3 nmol), 5 (3 μL, 6 nmol) and 7 (3 μL, 6 nmol) to the solution of RhB results in colour change from pink to colourless which is indicated by the gradual decrease in absorbance band at the RhB dye at 554 nm. The degradation of RhB takes 2.5 min in case of derivative 3 (Fig. 5), 4 min in case of derivative 5 (Fig. S24 in ESI†) and 9 min in case of derivative 7 (Fig. S30 in ESI†). The characteristic extinction peak of RhB at 554 nm peak is used as the reference extinction peak for the reaction kinetic study.Catalytic degradation of methylene blue, methyl orange and eriochrome black T was also carried out by using same procedure and results are summarized in Table S3, in ESI.†
3.5. Photocatalytic degradation of rhodamine B
The in situ generated AgNPs by aggregates of derivative 3 were also evaluated as photocatalysts for the degradation of RhB dye in the presence of visible light. The irradiation source was a 200 W tungsten filament lamp located inside the reactor. The RhB (2 mM) solution diluted 10 times in deionized water and was magnetically stirred for 30 min. in dark to establish adsorption/desorption equilibrium. The dye solution in the presence of in situ generated AgNPs catalyst 50 μL (50 nmol) was irradiated under visible light and small aliquots were withdrawn at regular intervals of time (after 1 hour) and UV-vis spectra were recorded. Under visible illumination, the dye is de-ethylated in a stepwise manner indicated by colour change of solution and blue shift in absorption spectra. After about 2 h, the main peak of RhB was blue shifted to 15 nm and about 60% degradation ratio was observed.4. Conclusion
Pentacenequinone, HPB and PDI based derivatives 3, 5 and 7 having aldehyde groups have been synthesized. All the derivatives formed aggregates in aqueous media and served as reactors and stabilizers for the preparation of AgNPs. Aggregates of derivatives 3 and 7 having flake like and porous morphology display high rate of formation of AgNPs, thus, showing the importance of morphology on rate of formation of AgNPs. Interestingly, in situ generated AgNPs of aggregates of derivatives 3, 5 and 7 exhibit higher catalytic efficiency for degradation of industrially important organic dyes.Acknowledgements
V. B. is thankful to CSIR [(ref. no. 02(0083)/12/EMR-II)]. M. K. is thankful to DRDO [(ref. no. ERIP/ER/1003922/M/01/1429)] and UGC [ref. no. 42-282/2013(SR)]. We are thankful to UGC for the UPE project.Notes and references
- H. Zhang, D. Chen, L. V. Xiaojun, Y. Wang, H. Chang and J. Li, Environ. Sci. Technol., 2010, 44, 1107–1111 CrossRef CAS PubMed.
- C. A. Martınez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105–145 CrossRef PubMed.
- F. C. Wu and R. L. Tseng, J. Hazard. Mater., 2008, 152, 1256–1267 CrossRef CAS PubMed.
- L. T. Gibson, Chem. Soc. Rev., 2014, 43, 5173–5182 RSC.
- N. Kannan and M. M. Sundaram, Dyes Pigm., 2001, 51, 25–40 CrossRef CAS.
- P. Pandit and S. Basu, Environ. Sci. Technol., 2004, 38, 2435–2442 CrossRef CAS.
- B. Neppolian, H. C. Choi, S. Sakthivel, B. Arabindoo and V. Murugesan, J. Hazard. Mater. B, 2002, 89, 303–317 CrossRef CAS.
- Y. Hsu, J. Chen, H. Yang and J. Chen, AIChE J., 2001, 47, 169–176 CrossRef CAS.
- C.-L. Yang and J. McGarrahan, J. Hazard. Mater. B, 2005, 127, 40–47 CrossRef CAS PubMed.
- Z. Xiong, L. L. Zhang, J. Ma and X. S. Zhao, Chem. Commun., 2010, 46, 6099–6101 RSC.
- Y. Pan, J. Wang, Y. Wang and Z. Wang, Macromol. Rapid Commun., 2014, 35, 635–641 CrossRef CAS PubMed.
- N. G. Bastús, F. Merkoçi, J. Piella and V. Puntes, Chem. Mater., 2014, 26, 2836–2846 CrossRef.
- L. Xu, X.-C. Wu and J.-J. Zhu, Nanotechnology, 2008, 19, 305603 CrossRef PubMed.
- Y. Wang, D. Zhao, W. Ma, C. Chen and J. Zhao, Environ. Sci. Technol., 2008, 42, 6173–6178 CrossRef CAS.
- W. Lu, X. Qin, H. Li, A. M. Asiri, A. O. Al-Youbi and X. Sun, Part. Part. Syst. Charact., 2013, 30, 67–71 CrossRef CAS.
- E. C. L. Ru and P. G. Etchegoin, Elsevier Science, 2008 Search PubMed.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669 CrossRef CAS PubMed.
- D. Yu and V.W.-W. Yam, J. Am. Chem. Soc., 2004, 126, 13200 CrossRef CAS PubMed.
- M. H. Rashid and T. K. Mandal, J. Phys. Chem. C, 2007, 111, 16750 CAS.
- R. Zong, X. Wang, S. Shi and Y. Zhu, Phys. Chem. Chem. Phys., 2014, 16, 4236–4241 RSC.
- G. Upert, F. Bouillére and H. Wennemers, Angew. Chem., 2012, 124, 4307–4310 CrossRef.
- I. Medina-Ramireza, S. Bashirb, Z. Luoc and J. L. Liu, Colloids Surf., B, 2009, 73, 185–191 CrossRef PubMed.
- R. R. Naik, S. E. Jones, C. J. Murray, J. C. McAuliffe, R. A. Vaia and M. O. stone, Adv. Funct. Mater., 2004, 14, 25–30 CrossRef CAS.
- G. S. Métraux and C. A. Mirkin, Adv. Mater., 2005, 17, 412–415 CrossRef.
- V. Bhalla, A. Gupta and M. Kumar, Chem. Commun., 2012, 48, 11862–11864 RSC.
- K. Sharma, S. Kaur, V. Bhalla, M. Kumar and A. Gupta, J. Mater. Chem. A, 2014, 2, 8369–8375 Search PubMed.
- K. Sharma, V. Bhalla and M. Kumar, RSC Adv., 2014, 4, 53795–53800 RSC.
- S. Pramanik, V. Bhalla and M. Kumar, Chem. Commun., 2014, 50, 13533–13536 RSC.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- V. Bhalla, S. Pramanik and M. Kumar, Chem. Commun., 2013, 49, 895–897 RSC.
- Q. Zhao, S. Zhang, Y. Liu, J. Mei, S. Chen, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 7387–7394 RSC.
- C. R. Swartz, S. R. Parkin, J. E. Bullock, J. E. Anthony, A. C. Mayer and G. G. Malliaras, Org. Lett., 2005, 7, 3163–3166 CrossRef CAS PubMed.
- R. M. Adhikari, B. K. Shah, S. S. Palayangoda and D. C. Neckers, Langmuir, 2009, 25, 2402–2406 CrossRef CAS PubMed.
- S. Basak, S. Bhattacharya, A. Datta and A. Banerjee, Chem.–Eur. J., 2014, 20, 5721–5726 CrossRef CAS PubMed.
- S. Agnihotri, S. Mukherji and S. Mukherji, RSC Adv., 2014, 4, 3974–3983 RSC.
- J. Sharma, N. K. Chaki, S. Mahima, R. G. Gonnade, I. S. Mulla and K. Vijayamohanan, J. Mater. Chem., 2004, 14, 970–975 RSC.
- S. Ghosh, V. S. Goudar, K. G. Padmalekha, S. V. Bhat, S. S. Indi and H. N. Vasan, RSC Adv., 2012, 2, 930–940 RSC.
- S. Goswami, S. Das, K. Aich, D. Sarkar, T. K. Mondal, C. K. Quah and H.-K. Fun, Dalton Trans., 2013, 15113–15119 RSC.
- M. Y. Han, C. H. Quek, W. Huang, C. H. Chew and L. M. Gan, Chem. Mater., 1999, 11, 1144–1147 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Mater., 2001, 13, 2313–2322 CrossRef CAS.
- A. Alahmad, M. Eleoui1, A. Falah and I. Alghoraibi, Physical Sciences Research International, 2013, 14, 89–96 Search PubMed.
- X. Sun, S. Dong and E. Wang, J. Colloid Interface Sci., 2005, 290, 130–133 CrossRef CAS PubMed.
- Y. Terazono, P. A. Liddell, V. Garg, G. Kodis, A. Brune, M. Hambourger, A. L. Moore, T. A. Moore and D. Gust, J. Porphyrins Phthalocyanines, 2005, 9, 706–723 CrossRef CAS.
- S. Vajiravelu, L. Ramunas, G. J. Vidas, G. Valentas, J. Vygintas and S. Valiyaveetqtil, J. Mater. Chem., 2009, 19, 4268–4275 RSC.
- A. S. Poyraz, C.-H. Kuo, S. Biswas, C. K. King'ondu and S. L. Suib, Nat. Commun., 2013, 4, 106–113 Search PubMed.
- A. J. Marenco, D. B. Pedersen, S. Wang, M. W. P. Petryk and H.-B. Kraatz, Analyst, 2009, 134, 2021–2027 RSC.
- M. Noel and K. I. Vasu, Cyclic Voltametry and The Frontiers of Electrochemistry, Oxford and IBH Publishing, New Delhi, India, 1990 Search PubMed.
- T. K. Sau and C. J. Murphy, Langmuir, 2004, 20, 6414–6420 CrossRef CAS PubMed.
- Z. S. Pillai and P. V. Kamat, J. Phys. Chem. B, 2004, 108, 945–951 CrossRef CAS.
- H. Li, H. Xia, D. Wang and X. Tao, Langmuir, 2013, 29, 5074–5079 CrossRef CAS PubMed.
- T. Chen, Y. Zheng, J.-M. Lin and G. Chen, J. Am. Soc. Mass Spectrom., 2008, 19, 997–1003 CrossRef CAS PubMed.
- For comparison Table S2 in in ESI.†.
- W. Zhang, X. Yang, Q. Zhu, K. Wang, J. Lu, M. Chen and Z. Yang, Ind. Eng. Chem. Res., 2014, 53, 16316–16323 CrossRef CAS.
- T. Wu, G. Liu, J. Zhao, H. Hidaka and N. Serpone, J. Phys. Chem. B, 1998, 102, 5845–5851 CrossRef CAS.
- T. Watanabe, T. Takizawa and K. Honda, J. Phys. Chem., 1977, 81, 1845–1851 CrossRef CAS.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Optical and spectroscopic data of derivatives 3, 5, 7. See DOI: 10.1039/c5ra02909k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |