
Synthesis, self-assembly, and catalytic activity of histidine-based structured lipopeptides for hydrolysis reactions in water†
Abstract
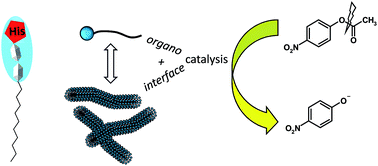
A new series of lipopeptides was designed to study their organocatalytic properties towards ester hydrolysis and the role of their self-assembled structures in catalysis. Synthesis of the catalysts was achieved by grafting fatty chains on tripeptides either at the C-terminal position or at the N-terminal extremity, affording amphiphilic character and self-assembling properties. Insertion of a histidine in the peptide sequence was chosen to bring about organocatalytic activity. Self-organization was evidenced first by the determination of critical aggregation concentrations and then by characterizing the aggregates formed by performing scattering techniques and microscopy. Variation of the structures (peptide sequence, hydrophobic character) led to the formation of various aggregates, from globular objects to fibers. All derivatives containing histidine presented a catalytic activity for the hydrolysis reaction of p-nitrophenyl acetate in aqueous solution. The influence of the self-organization on the catalysis was evidenced by showing different behaviors observed between the monomers and aggregates.
 Please wait while we load your content...
Please wait while we load your content...

