
Novel N-doped porous carbon microspheres containing oxygen and phosphorus for CO2 absorbent and metal-free electrocatalysts†
Abstract
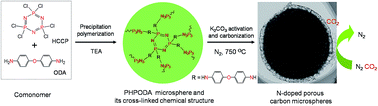
Novel N-doped porous carbon microspheres were synthesized facilely from polyphosphazene. These carbons exhibit a considerable performance for CO2 capture with high uptake, excellent selectivity, and good recyclability, and also show good electrocatalytic activity for oxygen reduction reaction.
 Please wait while we load your content...
Please wait while we load your content...

