
Nb-doped VOx/CeO2 catalyst for NH3-SCR of NOx at low temperatures†
Abstract
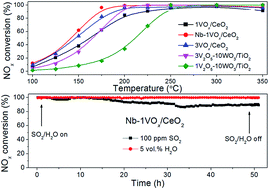
The promotion effect of Nb addition to VOx/CeO2 catalysts for the selective catalytic reduction of NOx by NH3 was fully investigated. VOx/CeO2 and NbOx doped VOx/CeO2 catalysts were characterized by N2 physisorption, XRD, H2-TPR and NH3-TPD. The results showed that the addition of Nb could significantly promote the SCR activity of the VOx/CeO2 catalyst, especially in the low temperature range. VOx/CeO2 with 30 wt% NbOx catalyst showed the best catalytic performance and better SO2/H2O tolerance than VOx/CeO2 catalyst. 30Nb-1VOx/CeO2 also exhibited higher NH3-SCR activity than 3V2O5–WO3/TiO2. Lower crystallinity, stronger redox capability and more Brønsted acid sites of the Nb-VOx/CeO2 catalyst were all responsible for its more excellent NH3-SCR performance. Based on kinetic experiments and in situ DRIFTS results, it was concluded that the Langmuir–Hinshelwood mechanism existed for selective catalytic reduction of NO over Nb-VOx/CeO2, in which adsorbed NOx species reacted with adsorbed NH3 to finally form N2 and H2O.
 Please wait while we load your content...
Please wait while we load your content...

