
Unusual crystallographic existence of a hydrated zinc(ii) bisulphate complex: experimental and theoretical observations†
Abstract
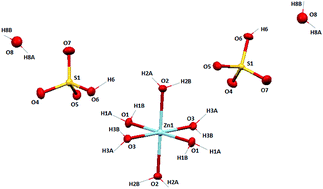
A unprecedented hydrated zinc(II) bisulphate complex, [Zn(H2O)6](HSO4·H2O)2 (1) has been synthesized along with a molecular ion salt, [4,4′-bipy-H+][NCS−] (2) (4,4′-bipy = 4,4′-bipyridine) and characterized by different spectroscopic tools including a single crystal X-ray diffraction study. From the X-ray crystal structure of 1, it is revealed that the Zn(II) ion is in a perfect octahedral geometry with a ZnO6 core and the molecule crystallizes in the P21/c space group. Self-assembly of the bisulphate anions with lattice water molecules pack to form a 12-membered cyclic cluster through strong intermolecular H-bonding (O–H⋯O–S). Again four cyclic tetramers interact with one another via H⋯O bonding to form a 20-membered macrocyclic bisulphate–water cluster and extends as a 2D network along the a axis. It presents a new mode of association of water molecules with bisulphate molecules. Density Function Theory (DFT) studies agree well with the experimental findings.
 Please wait while we load your content...
Please wait while we load your content...

