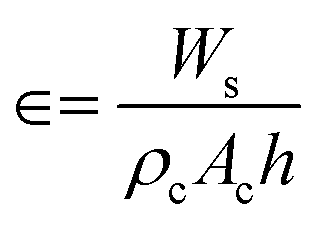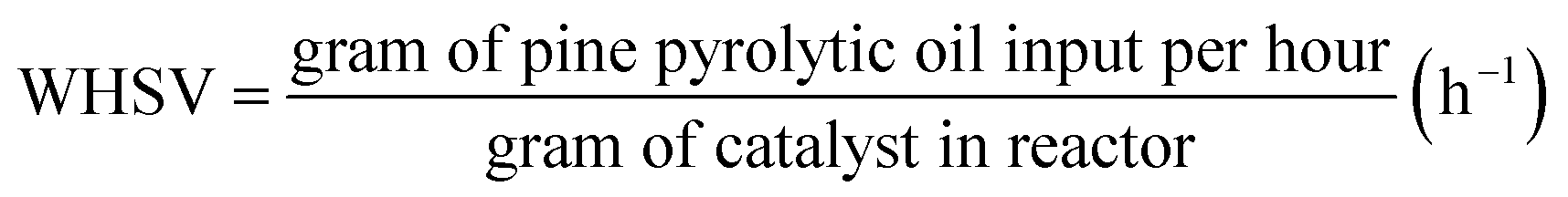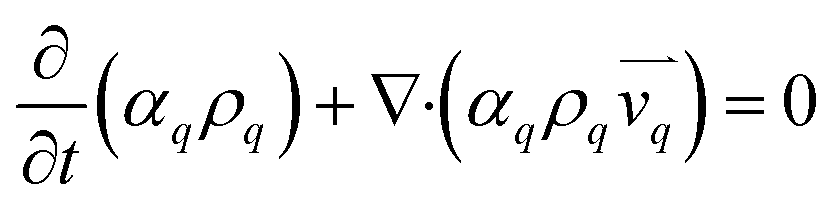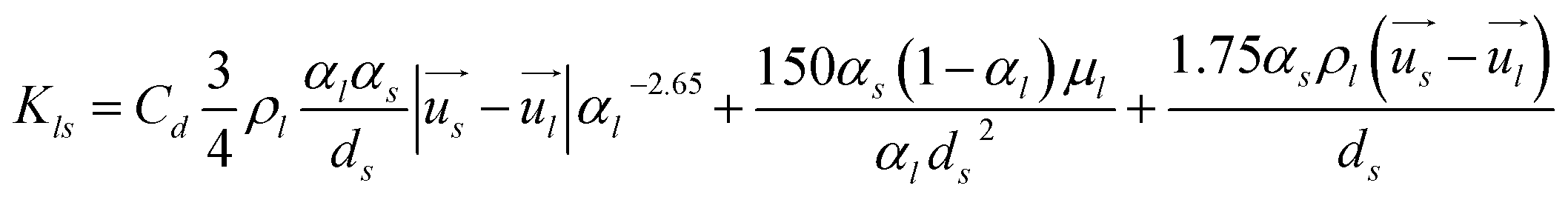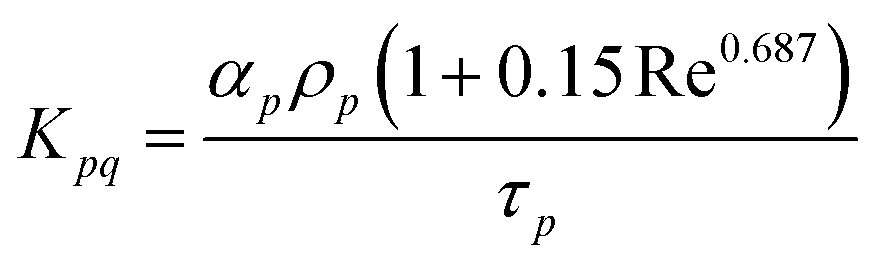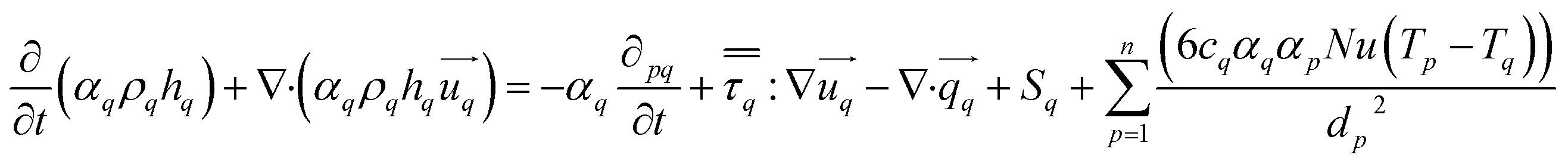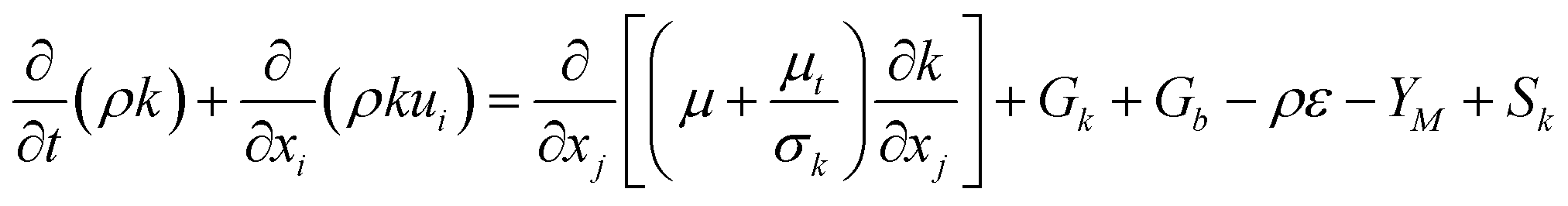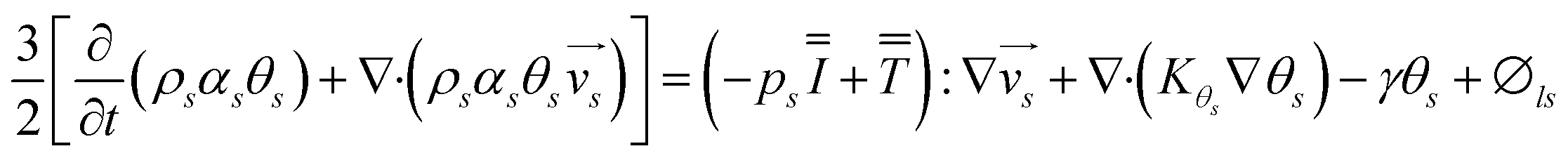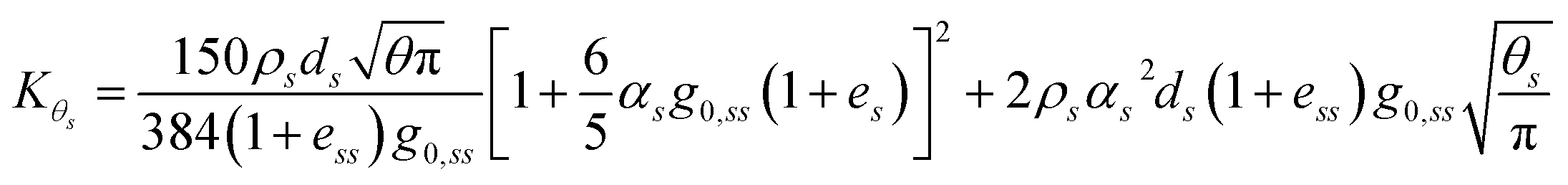DOI:
10.1039/C5RA02626A
(Paper)
RSC Adv., 2015,
5, 41855-41866
CFD simulations on the effect of catalysts on the hydrodeoxygenation of bio-oil
Received
10th February 2015
, Accepted 16th April 2015
First published on 16th April 2015
Abstract
Bio-oil derived from lignocellulose biomass is an emerging alternative resource to conventional fossil fuel. However, the as-obtained unprocessed bio oil is oxy-rich, has low pH and contains high moisture, which suppresses the heating value; thus, its mixing with conventional fuel is not compatible. Therefore, studies on the upgradation of bio oil using catalytic hydrodeoxygenation (HDO) have become prominent in recent years. This study presents computational fluid dynamics (CFD) based simulation results on the effect of catalysts (Pt/Al2O3, Ni–Mo/Al2O3, Co–Mo/Al2O3) on the upgradation of bio oil using a hydrodeoxygenation process in an ebullated bed reactor. These numerical simulations are performed using an Eulerian multiphase flow module that is available in a commercial CFD based solver, ANSYS Fluent 14.5. Prior to obtaining the new results, the present numerical solution methodology is validated by reproducing some of the experimental results on the upgradation of bio oil available in the literature. Furthermore, the influence of weight hourly space velocities (WHSVs), operating temperature, and pressure inside the reactor for the different catalysts on the performance of HDO for bio oil upgradation in an ebullated bed reactor are delineated. It is observed that the gaseous stream products are higher in the presence of Pt/Al2O3 catalyst; phenols are higher when Ni–Mo/Al2O3 is used, and higher aromatics are obtained with the Co–Mo/Al2O3 catalyst. Finally, a comparison among the mass fraction of the individual species of three phases with respect to different catalysts for various combinations of WHSV, temperature and pressure values are presented.
Introduction
Demand for energy is increasing globally and is expected to double in coming years due to population growth and various developments in society. The major source of energy generation to meet the present necessities is from fossil fuels. This energy generation, which results in the emission of CO2, leads to problems related to climate change such as global warming. Thus, it is a global challenge to counterbalance the environmental protection and generation of alternative sources of fuel to satisfy the demand. To address these challenges, mankind is stimulated to use renewable energies such as wind, solar, biomass and hydroelectricity, among which biofuels are emerging as a promising solution and alternative source of energy due to their sustainability and CO2 neutral resources. These bio fuels are derived from the biological carbon fixation and mainly result from biomass feed stocks. Some interesting facts about biomass feed stocks are that they are free from sulphur, nitrogen and ash; thus, the emissions are also free from SOx, NOx, and CO2. Because of their diversity, bio-fuels are classified into various sections and named as renewable advanced bio fuels or next generation sustainable fuels. This classification is majorly dependant on the type of feedstock, conversion technology, product formed and carbon source. Bioethanol and bio-diesel are first generation biofuels derived from biomass, whereas second generation biofuels are derived from lignocellulosic biomass. However, the major obstacle for these biofuels is the degradation of the biomass. Therefore, third generation biofuels are derived from microalgae and cyanobacteria. The bio oil that is obtained from the pyrolysis of lignocellulosic biomass is unstable and has a high water content, low pH, high viscosity, low heating value, and is highly corrosive. According to Oasmaa et al.,1 the produced bio-oil made of 300 different organic compounds mostly consists of (20–30 wt%) water, (15–30 wt%) lignin fragments, (10–20 wt%) aldehydes, (10–15 wt%) carboxylic acids, (5–10 wt%) carbohydrates, (2–5 wt%) phenols, (1–4 wt%) furfurals, (2–5 wt%) alcohols and (1–5 wt%) ketones. To overcome the deleterious properties of biomass pyrolysis oil, an upgrading process is required before its application. Currently, there are various techniques available to upgrade bio-oils into transportation fuels. They are catalytic hydrodeoxygenation (Furimsky2), zeolite upgrading (Adjaye and Bakhshi3), catalytic cracking (Hew et al.4), super critical technology (Tang et al.,5 Zhang et al.6) and emulsification (Bridgwater7).
Research activities on the upgradation of bio-oils using HDO started in the 1970's. The first review on the accomplishment of the upgradation process of bio-oil through HDO was successfully explained by Furimsky.2 Other pioneering work of Furimsky8 explained the chemistry, difficulties in determining rate constants, problems associated with the presence of oxygen, the growing concerns of upgrading coal and biomass derived fuels in detail. This led to a paradigm shift in research towards techniques pertaining to upgrading bio-oils. Oyama9 reported that the HDO process is similar to hydrodenitrogenation (HDN) but 10 times efficient than the later technique over vanadium nitride catalysts. Maggi and Delmon10 published a review, and discussed various aspects related to the catalytic chemistry, kinetics, and mechanisms of HDO reactions using various model oxygenated compounds inline with the review of Furimsky.2 Senol,11 Mahfud12 and Gutierrez et al.13 explained the process of removing oxygen from bio-oil using HDO under high pressure in the presence of suitable catalysts such as cobalt–molybdenum or nickel–molybdenum. Mahfud12 presented the reaction stoichiometry of the HDO process and reported that HDO is efficient in terms of carbon efficiency and saturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds and aromatic rings while removing oxygen in the presence of H2 and catalysts, resulting in the production of renewable liquid fuels such as gasoline and diesel. Furthermore, in extension to the earlier studies, Gutierrez et al.14 reported that upgrading bio oil by HDO requires relatively high pressures in the range of 7–20 MPa to convert some of the compounds of bio-oils that have a low HDO reactivity to O-free products. In addition, Elliott and Hart15 conducted semi batch HDO experiments using acetic acid and furfural to represent pyrolysis products from hemi-cellulose and cellulose, respectively, in the absence of a catalyst. The authors reported the formation of a solid polymeric material from furfural at 250 °C. Using Ru/C as the catalyst and acetic acid as the feed, they observed negligible conversions at low temperatures (<200 °C) and strong gas production at high temperatures (>250 °C). Their approach resulted in a reduction of the oxygen content from 41.3 wt% to 20–27.0 wt%.16–18 Wildschut et al.19 conducted HDO experiments in an autoclave using glucose and cellobiose as model compounds for the sugar fraction of pyrolysis oil. They concluded that during HDO of these model compounds using a ruthenium on carbon (Ru/C) catalyst, the catalytic hydrotreatment route is preferred over thermal decomposition, which leads to the formation of tar/solids (humins). The main products observed were polyols and gas products (mostly methane). Later Wildschut et al.20 found that there is no formation of benzene in the product on the HDO reaction of phenol over Ru/C catalyst. Recent results of Wildschut et al.,20 Li and Huber,21 and de Miguel Mercader et al.22 indicate that the HDO process removes oxygen under high pressures with a zeolite catalyst in the form of CO, CO2 and H2O. Furthermore, aqueous-phase reforming has been developed and tested for the bio-oil upgradation by Taarning et al.23 Another historical review of Mortensen et al.24 on the catalytic upgradation of bio-oil to engine fuels suggested two different paths for the upgradation process: hydrodeoxygenation and zeolite cracking. The author reported that the HDO process mostly occurs in the temperature range of 310–350 °C and pressures of 10–140 bar. They suggested a replacement of the catalyst suitable for HDO, which includes a sulphide catalyst and a noble catalyst with a base metal catalyst; this was supported by Wang et al.25 Moreover, HDO is found to be a suitable way to produce synthetic fuels of acceptable grade for the current infrastructure and also its usage as platform chemical to co-process in refinery units.22,26–30 Bridgwater7 presented a review on the fast pyrolysis of biomass and suggested that the bio refineries have the best possible scope for bio-fuel upgradation. Recently, Yaseen et al.31 experimentally studied the HDO of fast pyrolysis bio-oils from various feedstocks using carbon-supported catalysts. They concluded that the switch grass bio-oil performed the best over Pt/C catalyst in terms of hydrogen consumption efficiency, deoxygenation efficiency, and types of bio-oil upgraded compounds. The extensive work that has been undertaken over the past 25–30 years in the field of catalytic hydrotreating of biomass-derived liquids has been thoroughly reviewed by several researchers.32–36
C and CO bonds and aromatic rings while removing oxygen in the presence of H2 and catalysts, resulting in the production of renewable liquid fuels such as gasoline and diesel. Furthermore, in extension to the earlier studies, Gutierrez et al.14 reported that upgrading bio oil by HDO requires relatively high pressures in the range of 7–20 MPa to convert some of the compounds of bio-oils that have a low HDO reactivity to O-free products. In addition, Elliott and Hart15 conducted semi batch HDO experiments using acetic acid and furfural to represent pyrolysis products from hemi-cellulose and cellulose, respectively, in the absence of a catalyst. The authors reported the formation of a solid polymeric material from furfural at 250 °C. Using Ru/C as the catalyst and acetic acid as the feed, they observed negligible conversions at low temperatures (<200 °C) and strong gas production at high temperatures (>250 °C). Their approach resulted in a reduction of the oxygen content from 41.3 wt% to 20–27.0 wt%.16–18 Wildschut et al.19 conducted HDO experiments in an autoclave using glucose and cellobiose as model compounds for the sugar fraction of pyrolysis oil. They concluded that during HDO of these model compounds using a ruthenium on carbon (Ru/C) catalyst, the catalytic hydrotreatment route is preferred over thermal decomposition, which leads to the formation of tar/solids (humins). The main products observed were polyols and gas products (mostly methane). Later Wildschut et al.20 found that there is no formation of benzene in the product on the HDO reaction of phenol over Ru/C catalyst. Recent results of Wildschut et al.,20 Li and Huber,21 and de Miguel Mercader et al.22 indicate that the HDO process removes oxygen under high pressures with a zeolite catalyst in the form of CO, CO2 and H2O. Furthermore, aqueous-phase reforming has been developed and tested for the bio-oil upgradation by Taarning et al.23 Another historical review of Mortensen et al.24 on the catalytic upgradation of bio-oil to engine fuels suggested two different paths for the upgradation process: hydrodeoxygenation and zeolite cracking. The author reported that the HDO process mostly occurs in the temperature range of 310–350 °C and pressures of 10–140 bar. They suggested a replacement of the catalyst suitable for HDO, which includes a sulphide catalyst and a noble catalyst with a base metal catalyst; this was supported by Wang et al.25 Moreover, HDO is found to be a suitable way to produce synthetic fuels of acceptable grade for the current infrastructure and also its usage as platform chemical to co-process in refinery units.22,26–30 Bridgwater7 presented a review on the fast pyrolysis of biomass and suggested that the bio refineries have the best possible scope for bio-fuel upgradation. Recently, Yaseen et al.31 experimentally studied the HDO of fast pyrolysis bio-oils from various feedstocks using carbon-supported catalysts. They concluded that the switch grass bio-oil performed the best over Pt/C catalyst in terms of hydrogen consumption efficiency, deoxygenation efficiency, and types of bio-oil upgraded compounds. The extensive work that has been undertaken over the past 25–30 years in the field of catalytic hydrotreating of biomass-derived liquids has been thoroughly reviewed by several researchers.32–36
Finally, from the aforementioned extensive literature review, it can be concluded that several experimental studies on the upgradation of bio oil using HDO in the presence of suitable catalysts are available in the literature; however, analogous information on the basis of numerical studies is virtually non-existent. Therefore, this study aims to numerically investigate the performance of HDO process for the upgradation of bio oil in the presence of Pt/Al2O3, Ni–MO/Al2O3, Co–MO/Al2O3 catalysts over wide ranges of WHSV, temperature and pressure using a CFD approach.
Problem statement and mathematical formulation
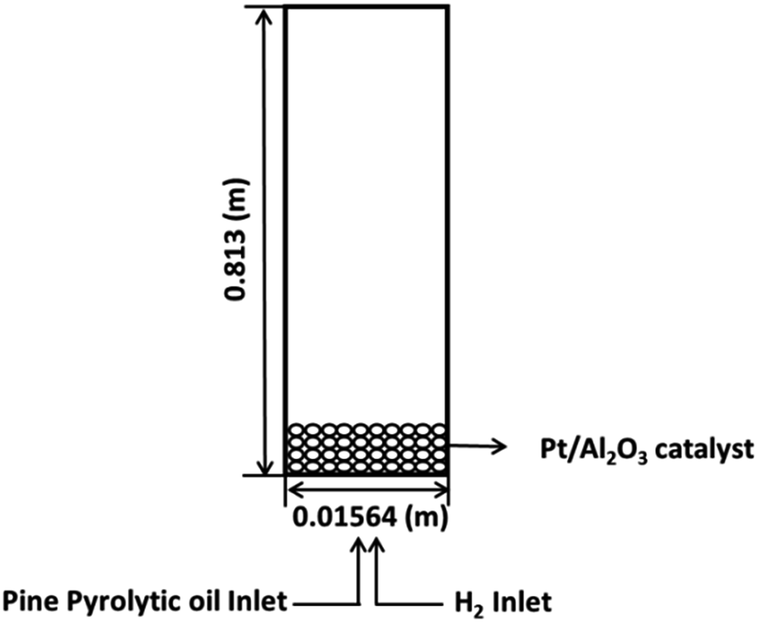
A schematic representation of the ebullated bed reactor used in the present simulation study is shown in Fig. 1. The height of the reactor is chosen to be 0.813 m and the diameter of the reactor is chosen to be 0.01564 m. The reactor is initially packed with catalyst particles up to 0.508 m of the maximum packing limit height. The conditions for the free board and catalyst bed are specified in terms of volume fraction. The catalyst volume fractions are obtained using the following equation:| | 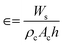 | (1) |
where Ws is the weight of the solid fed to the reactor, ρc is the density of the catalyst, Ac is the cross section area of the reactor, h is the packing height. The volume fraction of the catalyst in the bed is calculated to be 0.0286 in the case of Pt/Al2O3 catalyst, and it is 0.75 for Ni–Mo/Al2O3 and Co–Mo/Al2O3 catalysts. In other words, only a volume fraction of 0.0286 (out of the initial maximum packing height of 0.508 m) is occupied by the catalyst particles when Pt/Al2O3 catalyst is used; similarly, a volume fraction of 0.75 is occupied by the catalyst particles when Ni–Mo/Al2O3 and Co–Mo/Al2O3 catalysts are used. The pine pyrolytic oil consisting of various lumping groups along with hydrogen (H2) gas is introduced from the bottom of the reactor to pass through the catalyst bed. The thermo-physical properties of three phases used in the present simulations are listed in Table 1. The oil feed rate and its velocity is calculated based on the value of the weight hourly space velocity, which is given as follows:
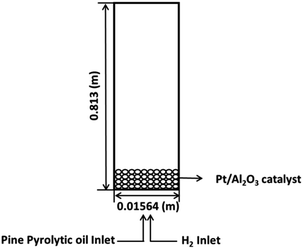 |
| | Fig. 1 Schematic representation of the ebullated bed reactor for bio-oil upgradation using HDO. | |
Table 1 Thermo-physical properties of the three phases (adopted from Stowe,48 Raal and Muhlbauer,49 Lin50)
| |
Compound |
ρ (kg m−3) |
μ (Pa s) |
C
p (J kg−1 K−1) |
K (W m−1 K−1) |
| Pine oil |
HNV |
841.15 |
0.0009 |
1833.81 |
0.127 |
| LNV |
679.5 |
0.0004 |
2223.19 |
0.140 |
| Phenols |
1030 |
0.1842 |
1430.00 |
0.190 |
| Aromatics |
880 |
0.0008 |
1699.84 |
0.131 |
| Alkane |
0.669 |
0.00001 |
2222 |
0.033 |
| Gas |
H2 (gas) |
0.8189 |
0.000008 |
14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 283 283 |
0.167 |
| Water vapour |
0.5542 |
0.000013 |
2014 |
0.0261 |
| Catalyst |
Pt/Al2O3 |
21450 |
0.000017 |
130 |
71.6 |
| Ni–Mo/Al2O3 |
829.75 |
0.000017 |
1360.71 |
0.186 |
| Co–Mo/Al2O3 |
829.75 |
0.000017 |
1243.47 |
0.2213 |
| Coke + ash |
375 |
1.206 |
850 |
0.2 |
For the gas phase inlet conditions, the minimum fluidization velocity of the gas phase is used in the present simulation studies. The bed region is initialized as a heterogeneous mixture of solid and gas phases, and the gas is fully occupied in the freeboard region.
To obtain the hydrodynamics and performance of the upgradation of bio oil, the following model equations along with appropriate reaction kinetics are solved simultaneously.
The continuity equation for all the three phases.37
| | 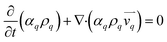 | (2) |
Fluid–fluid and fluid–solid momentum equation is given by Alder and Wainwright38
| | 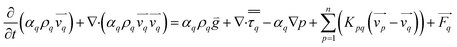 | (3) |
| | 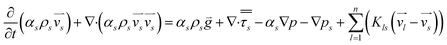 | (4) |
Interphase momentum exchange coefficient between liquid and solid phases.39–41
| | 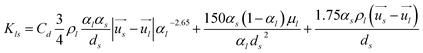 | (5) |
The drag between the solid and fluid wall is given by Schiller and Naumann42 as
| | 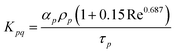 | (6) |
The energy conservation equation is given as
| | 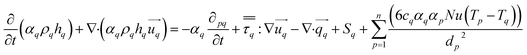 | (7) |
Fluid–fluid interaction is governed by Ranz and Marshall43 and fluid–solid by Gunn44
| | | Nup = 2.0 + 0.6Rep1/2Pr1/3 | (8) |
| | | Nus = (7 − 10αf + 5αf2)(1 + 0.7Res0.2Pr1/3) + (1.33 − 2.4αf + 1.2αf2)Res0.7 | (9) |
The turbulent kinetic energy (k) for the multiphase is governed by Launder and Spalding.45
| | 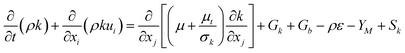 | (10) |
The dissipation rate (ε) of the turbulent kinetic energy for all the phases is also explained by Launder and Spalding.45
| | 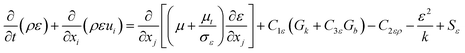 | (11) |
| | 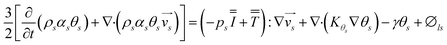 | (12) |
and finally the diffusion coefficient by Ding and Gidaspow.
46| | 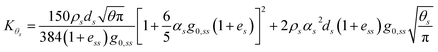 | (13) |
Lumped kinetic models
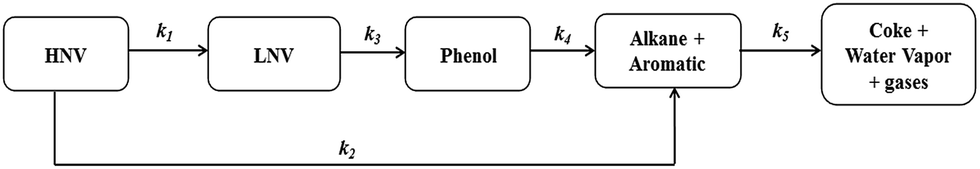
Because there are many species present in both pine pyrolytic oil and its hydrotreated products, the lumping of their constituents together with similar functional groups is a useful approach for studying reaction kinetics. Moreover, the lumped kinetic models give useful insights and a clear understanding to quantify the effects of process variables on product yields. In this study, five lumping kinetic model for the hydrodeoxygenation of pyrolytic bio-oil proposed by Sheu et al.47 is used, and the reaction pathway is shown in Fig. 2 and Table 2.
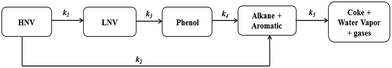 |
| | Fig. 2 Reaction pathways for the hydroprocessing of pine pyrolytic oil (Sheu et al.47). | |
Table 2 Lumped kinetic parameters of pine pyrolytic oil by Sheu et al.47
| |
HNV → LNV |
HNV → alkane + aromatic |
LNV → phenol |
Phenol → alkane + aromatic |
Alkane + aromatic → H2O + gases + coke |
|
Pt/Al
2
O
3
|
| Activation energy, Ea × 10−7 (J kmol−1) |
7.40 |
9.18 |
8.06 |
6.23 |
6.96 |
| Arrhenius constant, A (min−1) |
3860 |
75400 |
8300 |
950 |
4000 |
|
|
Ni–Mo/Al
2
O
3
|
| Activation energy, Ea × 10−7 (J kmol−1) |
8.22 |
10.58 |
9.04 |
6.84 |
7.49 |
| Arrhenius constant, A (min−1) |
8800 |
654000 |
30600 |
1920 |
16400 |
|
|
Co–Mo/Al
2
O
3
|
| Activation energy, Ea × 10−7 (J kmol−1) |
7.45 |
9.64 |
8.18 |
6.90 |
5.58 |
| Arrhenius constant, A (min−1) |
3500 |
218000 |
7700 |
3100 |
450 |
All these reactions are forward reactions, i.e., they are irreversible and their rate equations are given as follows:
| | | r2 = k1ρ1 − k2ρ2 − k3ρ2 | (15) |
| | | r4 = k2ρ2 + k4ρ3 − k5ρ4 | (17) |
where
ρ1,
ρ2,
ρ3, and
ρ4 are the densities of the heavy non-volatiles, light non volatiles, phenols and alkane aromatics, respectively. The reaction pathways follow the forward direction alone without any backward reactions, as shown in
Table 2 and
Fig. 2.
Numerical methodology
The aforementioned model equations for hydrodynamics and reaction kinetics are solved simultaneously using a turbulent flow module available in the commercial CFD software ANSYS Fluent 14.5 in double precision mode. This methodology employs a finite volume approach for flow solutions, which is beneficial for the local satisfaction of the conservation equations and for relatively coarse grid modelling. As shown in Fig. 1, the velocity inlet and pressure outlet boundary conditions are used for the present simulation studies because the realistic values promote numerical convergence. For wall boundaries, no slip boundary is applied. A pressure based solver is employed to solve phasic momentum equations, shared pressure, and phasic volume fraction equations in a segregated manner. The phase coupled semi-implicit method for pressure linked equations (PC-SIMPLE) algorithm is implemented, which is an extension of the SIMPLE algorithm developed for multiphase flows. In the PC-SIMPLE method, velocities are solved coupled by phases, yet in a segregated manner. A block algebraic multigrid scheme is then used to solve a vector equation of the velocity components of all phases simultaneously. For spatial discretization, a second order upwind scheme is chosen for the momentum equation, and QUICK scheme is chosen for volume fractions. The QUICK scheme is based on a weighted average of second-order upwind and central interpolations of the variable. The time step size used for simulation is in the order of 10−3. The structured quadrilateral grid is implemented using a hexahedral mesh of 12462 nodes. The post processing of the simulation results was performed using CFD post 14.5.
Results and discussion
Validation
The results on the upgradation of bio-oil using the HDO process through numerical approach are virtually non-existent and to the best of our knowledge, only Sheu et al.47 have reported numeric results on the upgradation of bio-oil using HDO.
Therefore, the present numerical solver is validated by comparing the present values of the mass fractions of the lumped species of the upgraded bio-oil phase with existing experimental results from Sheu et al.,47 and this is shown in Table 3. The present results are in close proximity with the existing literature values, which gave us the confidence to proceed further to check the effects of various catalysts over a wide range of temperatures, pressures and weight hourly space velocities.
Table 3 Validation of present results at T = 623 K, P = 8720 kPa and WHSV = 2 h−1 with the experimental results of Sheu et al.47
| Lumped fraction |
Unprocessed pyrolytic oil (wt%)47 |
Upgraded pyrolytic oil (wt%) |
| Experimental (Sheu et al.47) |
Present numerical results |
| HNV |
0.4932 |
0.2457 |
0.2102 |
| LNV |
0.3690 |
0.2941 |
0.4083 |
| Phenol |
0.1232 |
0.1063 |
0.1552 |
| Alkane + aromatic |
0.0146 |
0.1952 |
0.1962 |
| Coke + gas + H2O |
0 |
0.1587 |
0.000169 |
Volume fractions of upgraded pine oil, catalyst and H2 gas
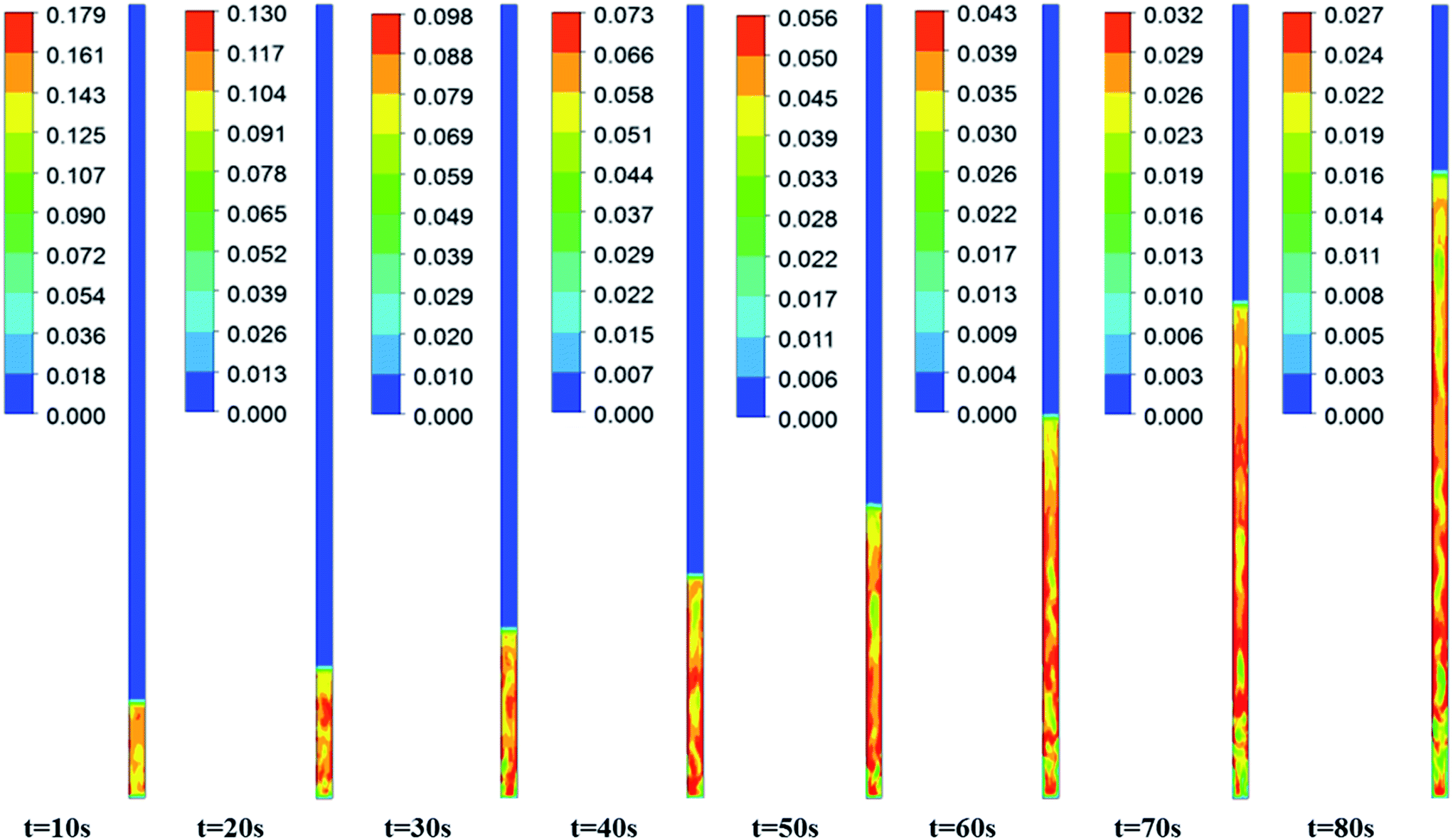
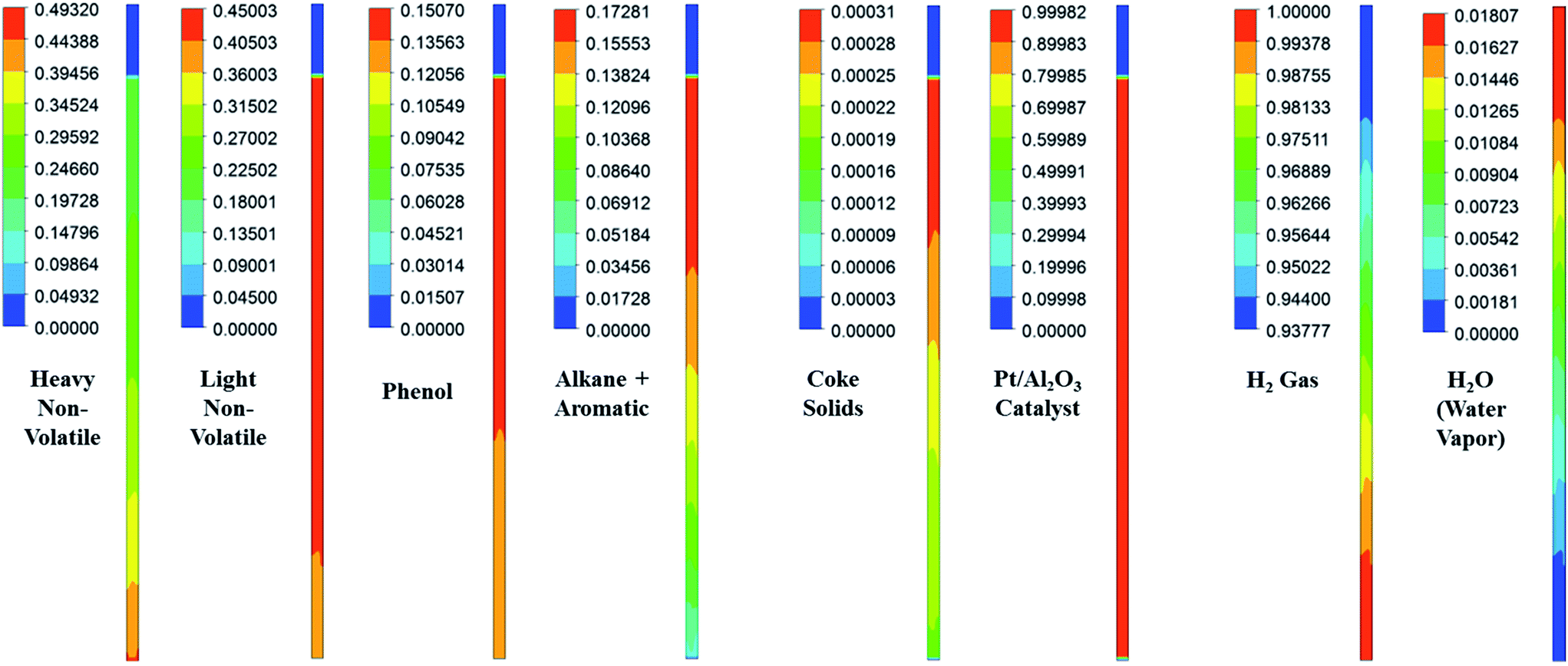
Fig. 3–5 show the prototype volume fraction images of all three phases (i.e., catalyst phase in Fig. 3, pyrolytic oil phase in Fig. 4 and H2 gas phase in Fig. 5) at T = 673 K and P = 8720 kPa at WHSV = 3 h−1 in the presence of Pt/Al2O3 catalyst. It can be noted from the volume fraction of all three phases that these phases expand with increasing time and reach a limiting maximum permissible height of 0.508 m of the bed. The volume fraction images of the catalyst phase (Fig. 3) indicate that the total volume of the catalyst remain constant though they distribute (expand) up to a bed height of 0.508 m. On the other hand, the volume fraction images of upgraded pine oil (Fig. 4) increases with increasing time, indicating the change in the composition of their lumped species and attain steady value at larger time values. The volume fraction images of H2 gas phase (Fig. 5) indicate that most of it occupies the free board space; however, a significant amount of H2 is also available in the bed region for the upgradation of bio-oil. Similarly, Fig. 6 denotes the steady mass fraction of the lumped species of the upgraded bio-oil obtained by the use of Pt/Al2O3 catalyst at WHSV = 3 h−1 at T = 673 K and P = 8720 kPa. The mass fraction images, shown in Fig. 6, have been obtained after the steady state has been reached, i.e., the bed expansion has ceased and no further change in the composition of upgrading bio-oil is observed.
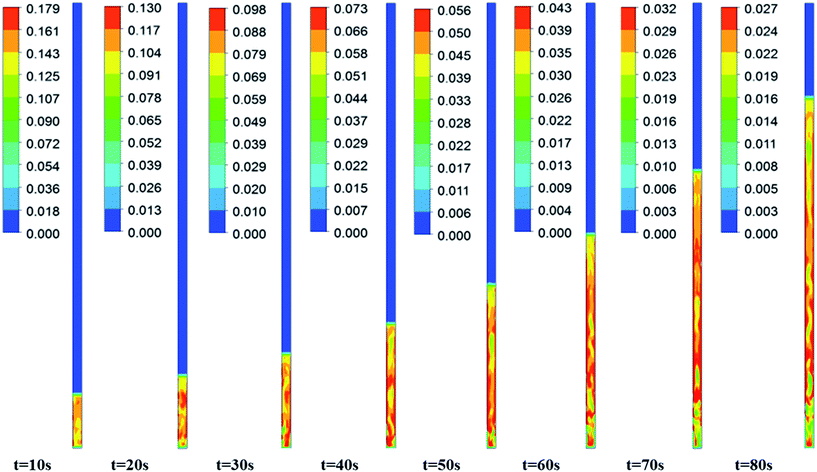 |
| | Fig. 3 Expansion of the Pt/Al2O3 catalyst bed at WHSV = 3 h−1, T = 673 K and P = 8720 kPa with increasing time. | |
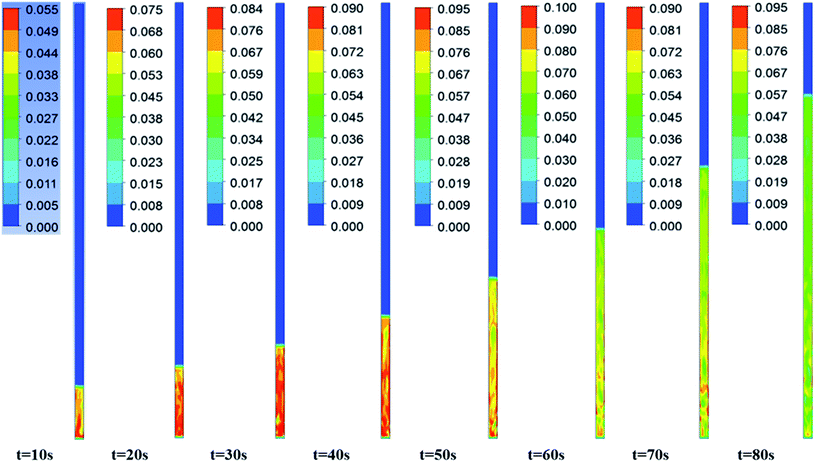 |
| | Fig. 4 Volume fraction images of the pine pyrolytic oil phase with increasing time at WHSV = 3 h−1, T = 673 K and 8720 kPa in the presence of Pt/Al2O3 catalyst. | |
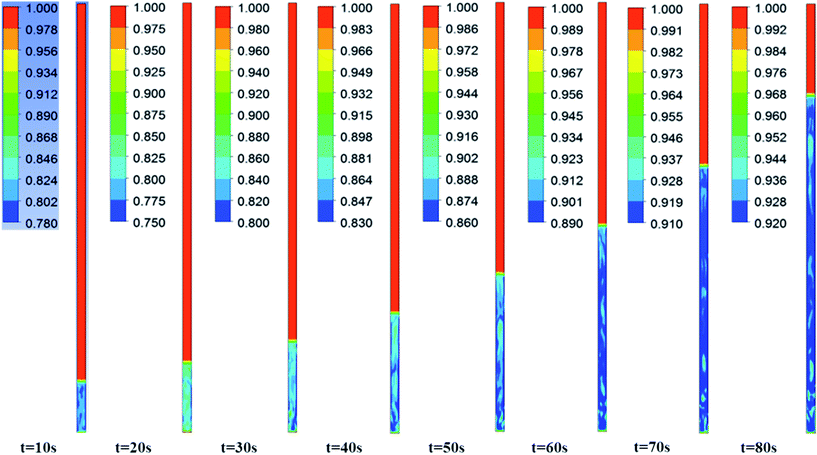 |
| | Fig. 5 Volume fraction of the H2 gas phase with increasing time at WHSV = 3 h−1, T = 673 K and 8720 kPa in the presence of Pt/Al2O3 catalyst. | |
 |
| | Fig. 6 Steady mass fraction images of the lumped species of upgraded pyrolytic oil and those of solid and gas/vapour phases in the presence of Pt/Al2O3 catalyst at WHSV = 3 h−1, T = 673 K and P = 8720 kPa. | |
It can be seen from Fig. 6 that all the lumped species of upgraded bio-oil are expanded within the maximum limit of bed height and their composition in the free board region is zero; however, the H2 gas escapes into the freeboard region. Moreover, the final steady mass fractions of HNV, LNV, phenols, alkanes and aromatics in this figure are consistent with the experimental mass fractions reported by Sheu et al.47 From these simulation results, it can be said that it is possible to almost completely overcome coke formation. Moreover, the water vapour (moisture) contents can be reduced to an almost negligible fraction (<2% vol) provided the experimental conditions are maintained exactly the same as in the simulations.
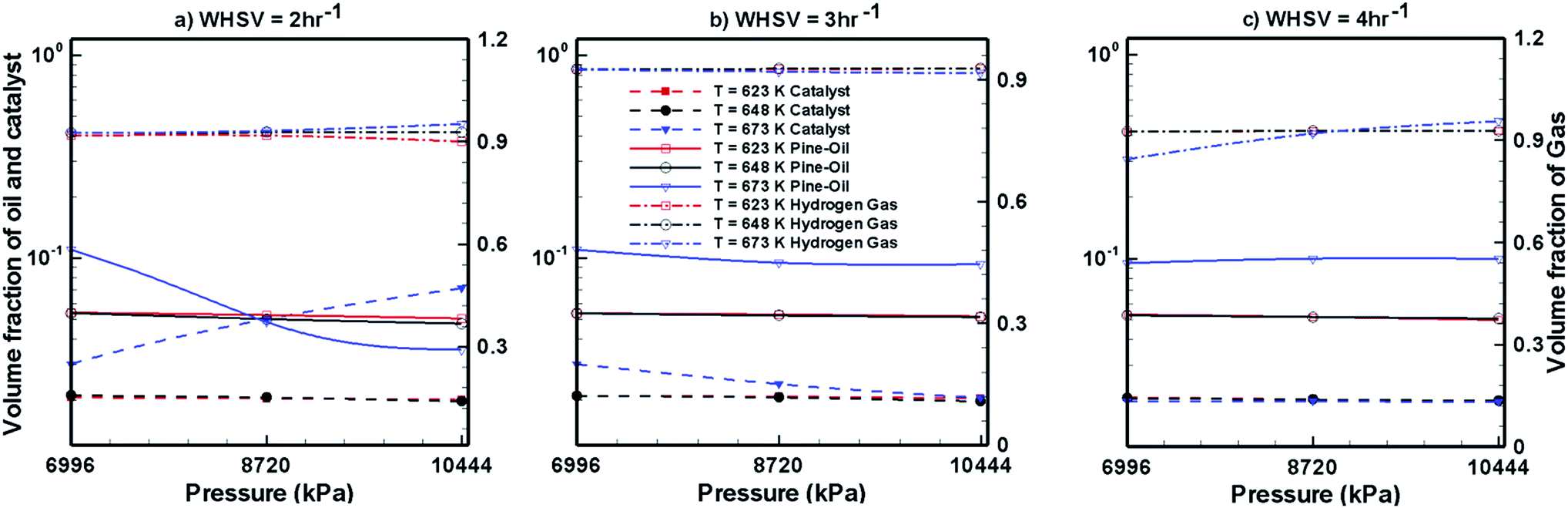
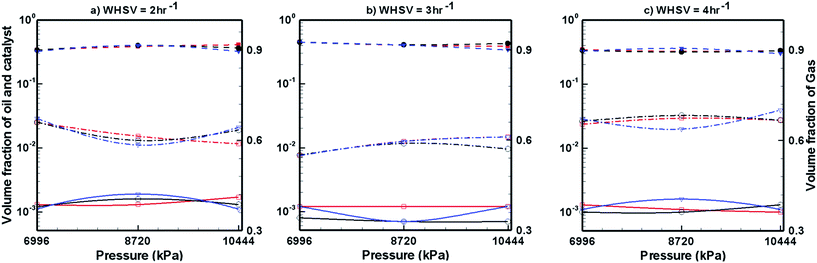
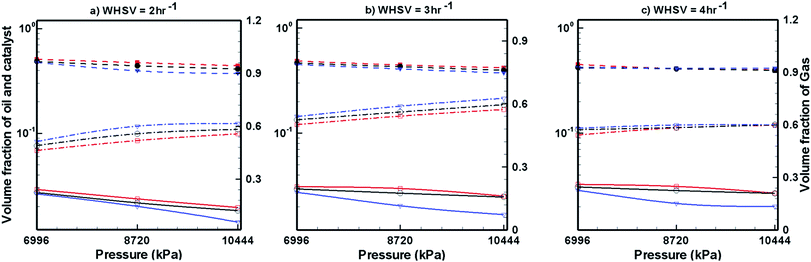
Fig. 7–9 show the effects of temperature, pressure and WHSV on the volume fraction of catalyst phase, H2 gas phase and upgraded oil phase in the presence of Pt/Al2O3 catalyst (Fig. 7), Ni–Mo/Al2O3 catalyst (Fig. 8) and Co–Mo/Al2O3 catalyst (Fig. 9). The line legends are same for Fig. 7–9; thus, they are shown in Fig. 8 only.
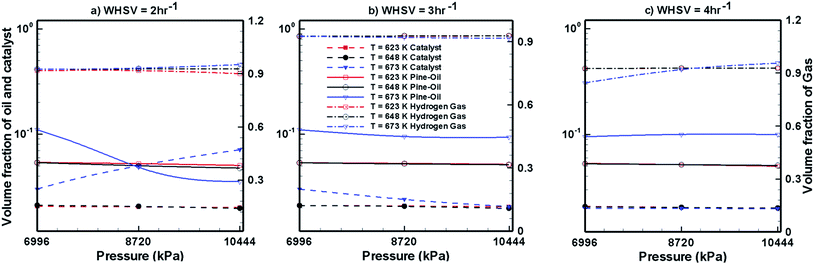 |
| | Fig. 7 Steady volume fractions of pine oil, H2 gas and Pt/Al2O3 catalyst at different temperatures and pressures. | |
 |
| | Fig. 8 Steady volume fractions of pine oil, H2 gas and Ni–Mo/Al2O3 catalyst at different temperatures and pressures. | |
 |
| | Fig. 9 Steady volume fractions of pine oil, H2 gas and Co–Mo/Al2O3 catalyst at different temperatures and pressures. | |
In Fig. 7–9, the left y-axis depicts the corresponding volume fraction values of pine oil and the catalyst, whereas the right y-axis indicates the volume fraction values of the H2 gas phase. It should be noted that the values of volume fraction of the three phases presented in Fig. 7–9 are steady state values, i.e., obtained after the bed has attained steady state by expanding up to the maximum attainable bed height. These volume fraction values also indicate that there is no further change in their values with increasing time. Fig. 7 shows the variations in the volume fraction of upgraded oil, H2 gas and Pt/Al2O3 catalyst for different values of WHSV, temperature and pressure. The pine oil volume fraction shows a mixed trend with respect to temperature and pressure at WHSV = 2 h−1 (Fig. 7a); however, at WHSV = 3 h−1 (Fig. 7b) and WHSV = 4 h−1 (Fig. 7c), it increases with increase in the temperature to T = 673 K. However, at a fixed temperature, the volume fraction of pine oil is almost unaffected by WHSV and pressure.
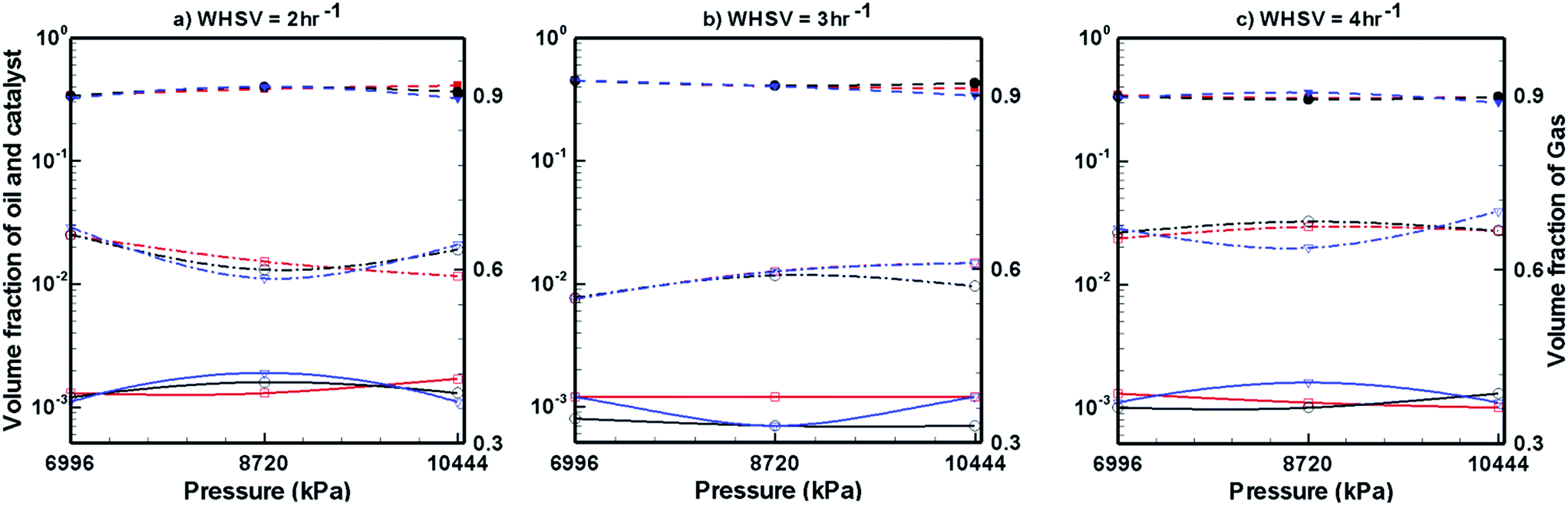
In the case of Ni–Mo/Al2O3 catalyst, the variation in catalyst expansion behaviour is almost negligible with the change in WHSV, temperature and pressure (Fig. 8). For the WHSV value of 2 h−1 (Fig. 8a), the steady volume fraction of H2 gas slightly decreases with pressure at T = 623 K, whereas at other temperatures, the volume fraction of H2 gas shows a mixed trend with increasing pressure. On increasing the WHSV value to 3 h−1 (Fig. 8b), the volume fraction of H2 gas at P = 6996 kPa is unaffected by the temperature; however, as the pressure increases to P = 8720 and 10444 kPa, mixed variations in the volume fraction of H2 gas are seen with increasing temperature. On further increasing the WHSV to 4 h−1 (Fig. 8c), the volume fraction of H2 gas at a given temperature and pressure increased in comparison to the case of WHSV = 3 h−1 (Fig. 8b). However, the volume fraction of H2 gas shows mixed variations with respect to temperature and pressure in the case of WHSV = 4 h−1. In the case of pine oil, the variations in their volume fraction with respect to temperature, pressure and WHSV values are very small and mixed behaviour is observed against changes in the operating conditions.
Fig. 9 shows the volume fraction of upgraded bio-oil, H2 gas and Co–Mo/Al2O3 catalyst for different WHSV, temperature and pressure values. The expansion of the catalyst bed is almost unaffected by WHSV, temperature and pressure. The volume fraction of H2 gas slightly increases with increasing pressure and temperature; however, mixed trends of H2 gas are seen with respect to the WHSV values. The volume fraction of pine oil decreases with increasing pressure and temperature; however, it increases with increasing WHSV values when Co–Mo/Al2O3 catalyst is used. Finally, by comparing the performance of all the catalysts (Fig. 7–9), it can be seen that Pt/Al2O3 produces a larger fraction of upgraded pine oil, whereas Ni–Mo/Al2O3 produces a smaller volume fraction of the upgraded bio-oil.
Mass fraction of lumped species of upgraded bio oil
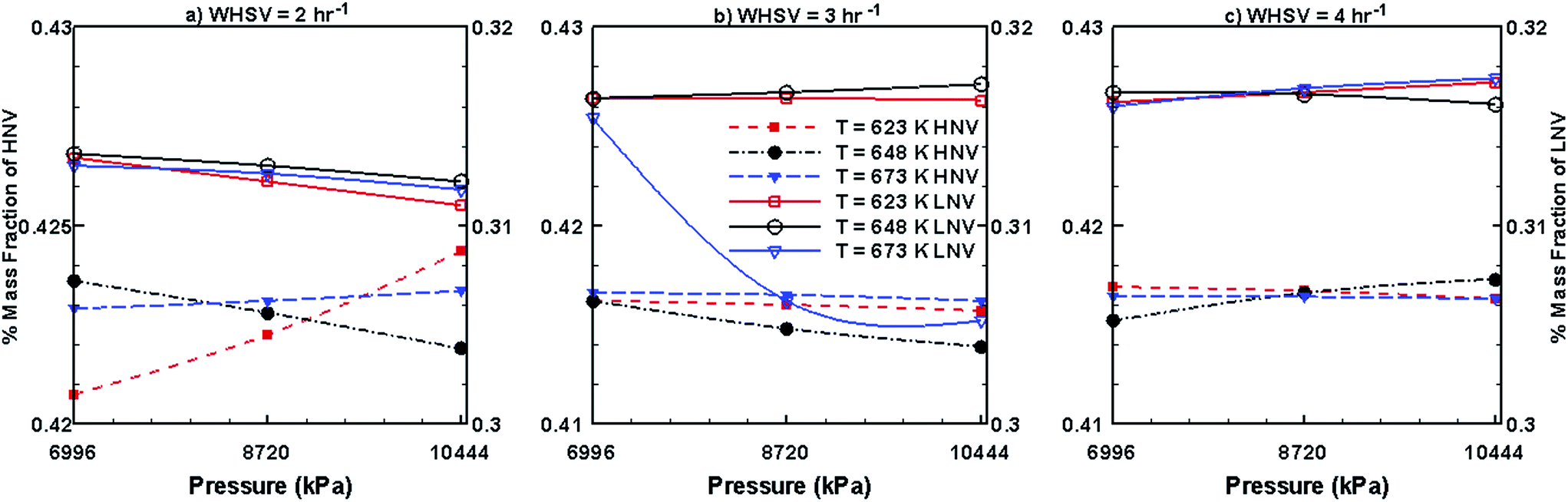
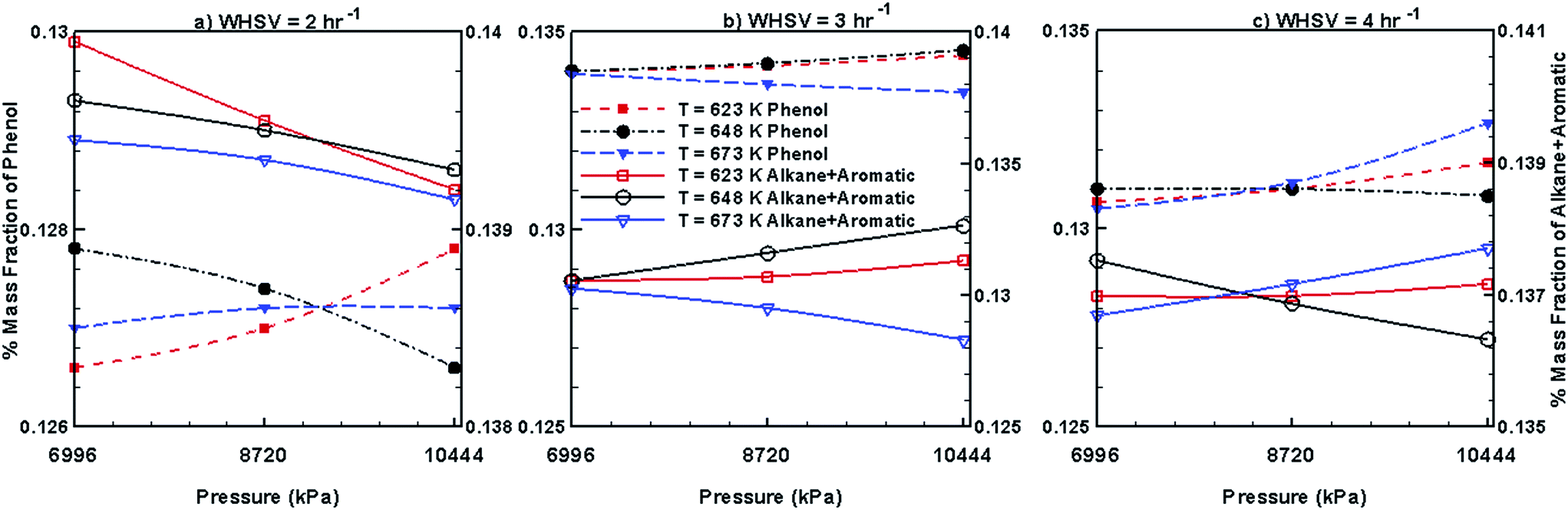
Fig. 10 and 11 show the steady mass fraction values of the lumped species of upgraded bio-oil by HDO process in the presence of Pt/Al2O3 catalyst for the different combinations of WHSV, temperature and pressure. The mass fraction values reported in these figures are obtained when the bed has reached pseudo steady state. Fig. 10 shows the steady mass fractions of lumped HNV and LNV in the presence of Pt/Al2O3 catalyst. At WHSV = 2 h−1, the steady mass fraction of LNV decreases slightly with increasing pressure and decreasing temperature. At WHSV = 3 h−1, the variation in the mass fraction of LNV is negligible with changes in temperature and pressure. At WHSV = 4 h−1, mixed variations in the steady state mass fractions of LNV are observed with respect to temperature and pressure. The mass fractions of HNV at WHSV = 2 h−1 (Fig. 10a) and WHSV = 4 h−1 (Fig. 10c) show a mixed trend with respect to temperature and pressure; however, at WHSV = 3 h−1 (Fig. 10b), at a given temperature, the mass fraction of HNV slightly decreases with increasing pressure. The variation in the mass fraction of HNV at WHSV = 3 h−1 (Fig. 10b) and a fixed value of pressure shows a mixed trend with respect to temperature. From Fig. 11a, it can be seen that the mass fractions of phenols, alkanes and aromatics show a mixed trend with respect to the WHSV, temperature and pressure.
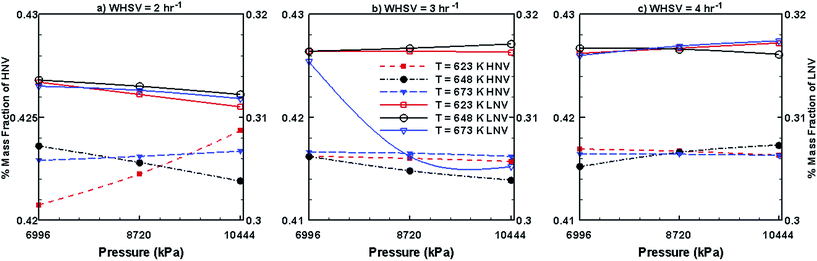 |
| | Fig. 10 Mass fractions of HNV and LNV obtained by upgrading pine oil in the presence of Pt/Al2O3. | |
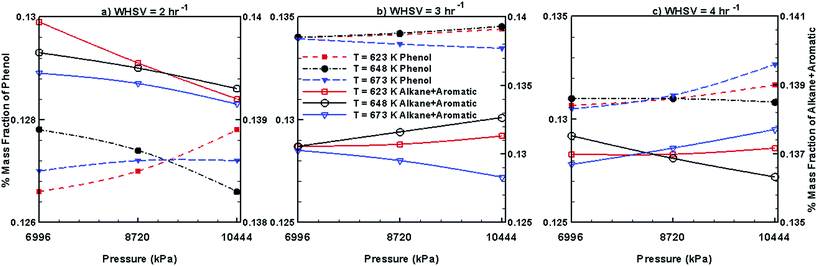 |
| | Fig. 11 Mass fractions of phenols, alkanes and aromatics obtained by upgrading pine oil in the presence of Pt/Al2O3. | |
Fig. 12 shows the variations in mass fraction values of lumped HNV and LNV species in the upgraded bio-oil by the HDO process in the presence of Ni–Mo/Al2O3 catalyst, and the mixed trends can be seen here too with respect to WHSV, temperature and pressure. However, compared to the Pt/Al2O3 catalyst, (Fig. 10) the mass fractions of HNV and LNV obtained by the use of Ni–Mo/Al2O3 has substantially decreased for fixed combinations of WHSV, temperature and pressure.
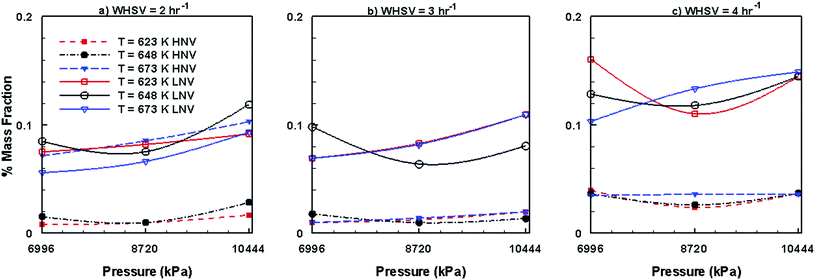 |
| | Fig. 12 Mass fractions of HNV and LNV obtained by upgrading pine oil in the presence of Ni–Mo/Al2O3. | |
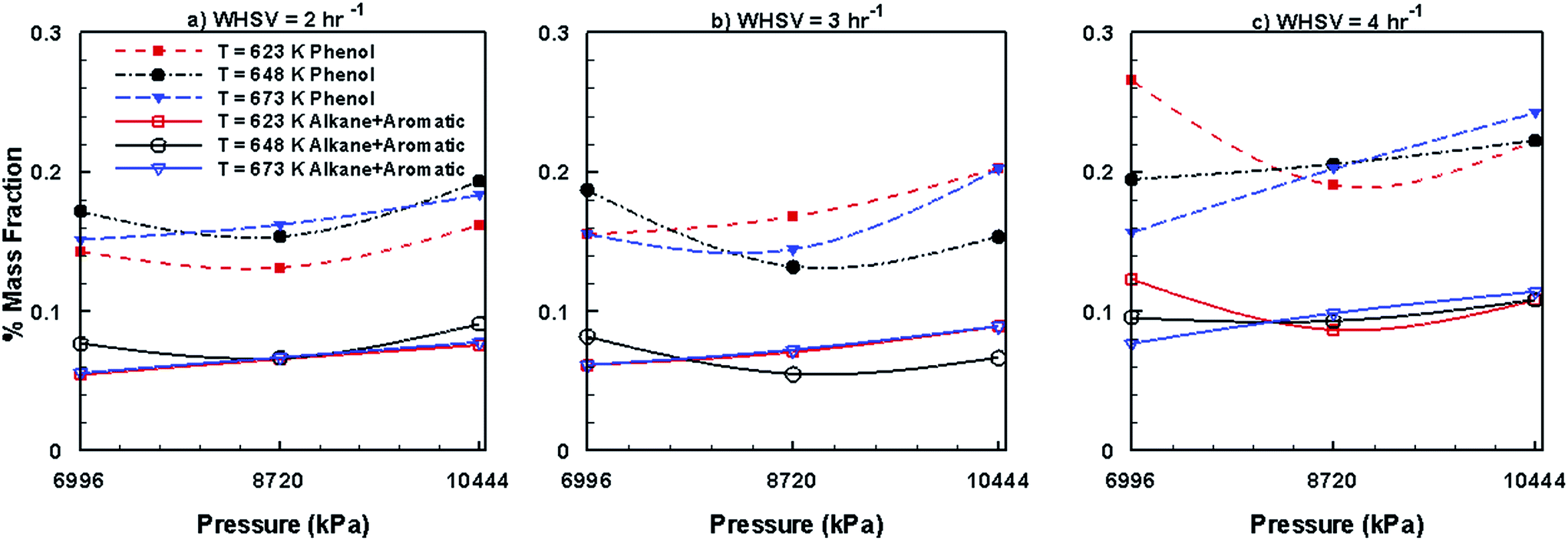
Fig. 13 shows the variations in the steady mass fractions of lumped phenols, alkanes and aromatics obtained by the HDO of bio-oil in the presence of Ni–Mo/Al2O3, and mixed trends are seen for changes in WHSV, temperature and pressure. However, compared to Pt/Al2O3 (Fig. 11), Ni–Mo/Al2O3 catalyst (Fig. 13) produces larger mass fractions of phenols but smaller fractions of alkanes and aromatics.
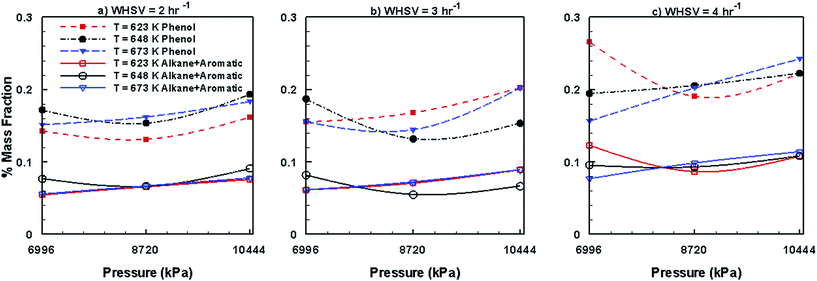 |
| | Fig. 13 Mass fractions of phenols, alkanes and aromatics obtained by upgrading pine oil in the presence of Ni–Mo/Al2O3. | |
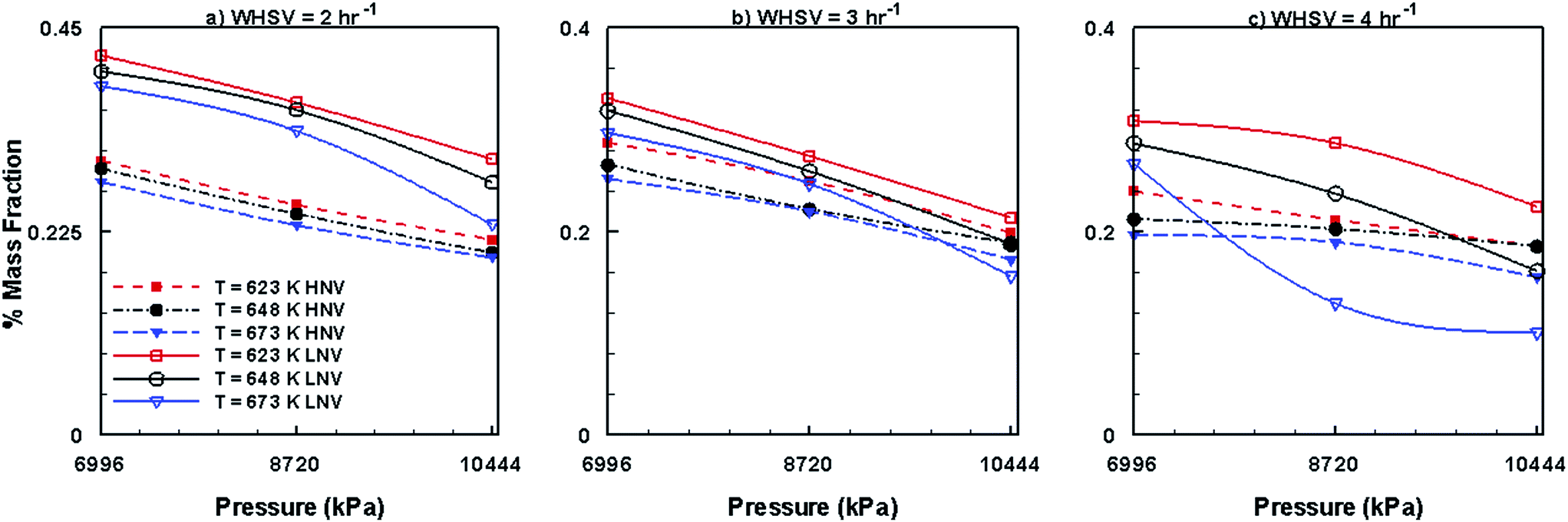
Fig. 14 shows the mass fractions of HNV and LNV obtained by the use of Co–Mo/Al2O3 catalyst. It can be seen from this figure that the mass fractions of both HNV and LNV decrease with increasing pressure, temperature and WHSV. In comparison to other catalysts, Ni–Mo/Al2O3 produces small fractions of HNV and LNV followed by Pt/Al2O3 catalyst and Co–Mo/Al2O3 catalyst producing larger fractions of HNV and LNV. Fig. 15 presents the mass fractions of phenols, alkanes and aromatics obtained by the use of Co–Mo/Al2O3 catalyst. It can be seen from Fig. 15 that the formation of phenols by the use of Co–Mo/Al2O3 is almost unaffected by temperature, pressure and WHSV values; however, the fractions of alkanes and aromatics increase with increasing pressure, temperature and WHSV. On comparing with other two catalysts, Co–Mo/Al2O3 catalyst produces larger fractions of alkanes and aromatics followed by Pt/Al2O3 and Ni–Mo/Al2O3, producing smaller fractions of alkanes and aromatics.
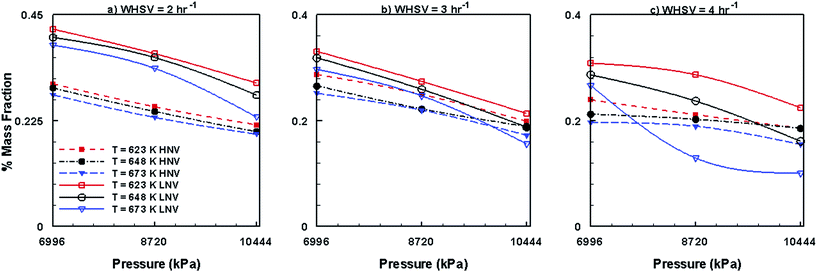 |
| | Fig. 14 Mass fractions of HNV and LNV obtained by upgrading pine oil in the presence of Co–Mo/Al2O3. | |
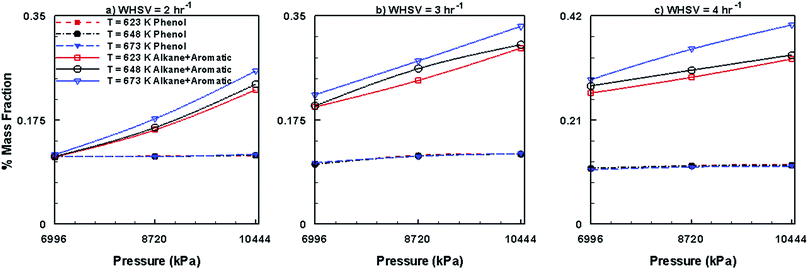 |
| | Fig. 15 Mass fractions of phenols, alkanes and aromatics obtained by upgrading pine oil in the presence of Co–Mo/Al2O3. | |
Conclusions
This numerical work presents the advancement of suitable catalysts for the upgradation of bio-oil through a HDO process in an ebullated bed reactor at temperatures running between 623 K ≤ T ≤ 673 K, pressure ranges between 6996 kPa ≤ P ≤ 10443 kPa and WHSVs varying between 2 ≤ WHSV (h−1) ≤ 4. The consequences of this study demonstrate some important and significant behaviour of the three phases under the influence of three different catalysts, namely, alumina supported platinum, Co–Mo, and Ni–Mo catalysts. Some of the key findings of this study include the fact that the gas volume fraction is higher in the case of Pt/Al2O3 catalyst. The amount of phenol formation during the upgradation process is significant and effective in the case of Ni–Mo/Al2O3 catalyst compared to the other catalysts. The amount of aromatic formation is larger by the use of Co–Mo/Al2O3 catalyst in comparison with the other catalysts. The higher values of the volume and the mass fractions of upgraded lumped species are obtained at low WHSV values and high temperatures and pressures.
Acknowledgements
Sai Gu gratefully acknowledges the financial support received from UK EPSRC Grant EP/K036548/1 and FP7 Marie Curie iComFluid Project Grant 312261. ARK Gollakota is very thankful to Dr P. Ranganathan (Cranfield University, UK) for his advice in the beginning stages of this work.
References
-
A. Oasmaa, D. Meier and A. Bridgwater, Fast Pyrolysis of Biomass: A Handbook, CPL Press, Newbury, UK, 2002, vol. 1 Search PubMed.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147 CrossRef CAS.
- J. D. Adjaye and N. N. Bakhshi, Biomass Bioenergy, 1995, 8, 265–277 CrossRef CAS.
- K. L. Hew, A. M. Tamidi, S. Yusup, K. T. Lee and M. M. Ahmad, Bioresour. Technol., 2010, 101, 8855–8858 CrossRef CAS PubMed.
- Z. Tang, L. Qiang, Z. Ying and G. Qingxiang, Ind. Eng. Chem. Res., 2009, 48, 6923–6929 CrossRef CAS.
- Y. Zhang, W. Li, S. Zhang, Q. Xu and Y. Yan, Chem. Eng. Technol., 2012, 35, 302–308 CrossRef CAS PubMed.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS PubMed.
- E. Furimsky, Catal. Rev.: Sci. Eng., 1983, 25, 421 CAS.
-
S. T. Oyama, The Chemistry of Transition Metal Carbides and Nitrides, Blackie Academic and Professional, Springer, Glasgow, 1996 Search PubMed.
- R. Maggi and B. Delmon, Stud. Surf. Sci. Catal., 1997, 106, 99 CAS.
-
O. Senol, PhD thesis, Helsinki University of Technology, 2007.
-
F. H. Mahfud, PhD thesis, University of Gronigen, 2007.
-
A. Gutierrez, M. E. Domine and Y. Solantausta, Co-processing of upgraded bio-liquids in standard refinery units-fundamentals, 15th European Biomass Conference and Exhibition, Berlin, 7–11 May 2007 Search PubMed.
- A. Gutierrez, R. K. Kaila, M. L. Honkela and R. Siloor, Catal. Today, 2009, 147, 239–246 CrossRef CAS PubMed.
- D. C. Elliott and T. R. Hart, Energy Fuels, 2009, 23, 631–637 CrossRef CAS.
-
D. C. Elliott and E. G. Baker, Process of Upgrading Biomass Pyrolyzates, US Pat., 4795851, 1989.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- D. C. Elliott, G. G. Neuenschwander and T. R. Hart, ACS Sustainable Chem. Eng., 2013, 1, 389–392 CrossRef CAS.
- J. Wildschut, I. M. Cabrere and H. J. Heeres, Ind. Eng. Chem. Res., 2009, 48, 10324–10334 CrossRef CAS.
- J. Wildschut, M. Iqbal, F. H. Mahfud, I. Melian Cabrera, R. H. Venderborsch and H. J. Herres, Energy Environ. Sci., 2010, 3, 962–970 CAS.
- N. Li and G. W. Huber, J. Catal., 2010, 270, 48–59 CrossRef CAS PubMed.
- F. de Miguel Mercader, M. J. Groeneveld, S. R. A. Kersten, N. W. J. Way, C. J. Schaverien and J. A. Hogendoorn, Appl. Catal., B, 2010, 96, 57–66 CrossRef CAS PubMed.
- E. Taarning, C. M. Osmundsen, X. Yang, B. Voss, S. I. Andersen and C. H. Chritensen, Energy Environ. Sci., 2011, 4, 793–804 CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- Y. Wang, Y. Fang, T. He, H. Hu and J. Wu, Catal. Commun., 2011, 12, 1201–1225 CrossRef CAS PubMed.
- W. Baldauf, U. Balfanz and M. Rupp, Biomass Bioenergy, 1994, 7, 237–244 CrossRef CAS.
- G. Fogassy, N. Thegarid, Y. Schuurman and C. Mirodatos, Green Chem., 2012, 14, 1367–1371 RSC.
-
E. G. Baker and D. C. Elliott, Method of Upgrading Oils Containing Hydroxyaromatic Hydrocarbon Compounds to Highly Aromatic Gasoline, US Pat., 5180868, 1993.
- T. Choudhary and C. Phillips, Appl. Catal., A, 2011, 397, 1–12 CrossRef CAS PubMed.
- R. H. Venderbosch, A. R. Ardiyanti, J. Wildschut, A. Oasmaa and H. J. Heeres, J. Chem. Technol. Biotechnol., 2010, 85, 674–686 CrossRef CAS PubMed.
- E. Yaseen, C. A. Mullen, A. L. M. T. Pighinelli and A. A. Boateng, Fuel Process. Technol., 2014, 123, 11–18 CrossRef PubMed.
- J. P. Diebold and J. W. Scahill, Energy Prog., 1988, 8, 59–65 CAS.
- E. M. Ryymin, M. L. Honkela, T. R. Viljava and A. O. I. Krause, Appl. Catal., A, 2010, 389, 114–121 CrossRef CAS PubMed.
- C. Zhao, S. Kasakov, J. He and J. A. Lercher, J. Catal., 2012, 296, 12–23 CrossRef CAS PubMed.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Appl. Catal., A, 2011, 391, 305–310 CrossRef CAS PubMed.
- Y. Zhao, L. Deng, B. Liao, Y. Fu and Q. Xiang Guo, Energy Fuels, 2010, 24, 5735–5740 CrossRef CAS.
-
FLUENT Inc., FLUENT User's Guide, 2006 Search PubMed.
- J. Alder and T. E. Wainwright, J. Chem. Phys., 1960, 33, 1439–1451 CrossRef PubMed.
-
D. Gidaspow, R. Bezburuah and J. Ding, Proceedings of the seventh Engineering Foundation Conference on Fluidization, Australia, 1992 Search PubMed.
- C. Y. Chen and Y. H. Hu, Chem. Eng. Prog., Symp. Ser., 1966, 62, 100–111 Search PubMed.
- S. Ergun, Chem. Eng. Prog., 1952, 48, 89–94 CAS.
- L. Schiller and Z. Naumann, Z. Ver. Dtsch. Ing., 1933, 77, 318–320 Search PubMed.
- W. E. Ranz and W. R. Marshall, Chem. Eng. Prog., 1952, 48, 141–146 CAS.
- D. J. Gunn, Int. J. Heat Mass Transfer, 1978, 21, 467–478 CrossRef.
-
B. E. Launder and D. B. Spalding, Lectures in mathematical models of turbulence, Academic Press, London and New York, 1972 Search PubMed.
- J. Ding and D. Gidaspow, AIChE J., 1990, 36, 523–528 CrossRef CAS PubMed.
- Y. H. E. Sheu, R. G. Anthony and E. J. Soltes, Fuel Process. Technol., 1988, 19, 31–50 CrossRef CAS.
-
L. R. Stowe, Method of Conversion of Heavy Hydrocarbon Feedstocks, US Pat., 5547563, 1996.
-
D. J. Raal and A. Muhlbauer, Phase Equilibria: Measurement and Computation, Taylor and Francis, Washington, USA, 1997 Search PubMed.
-
S. C. Lin, Hydrocarbons via catalytic hydrogen treatment of wood pyrolytic oil, PhD thesis, A&M University, Texas, 1981.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a