
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of CaCO3@C yolk–shell particles for CO2 adsorption†
Yash
Boyjoo
a,
Kelly
Merigot
b,
Jean-François
Lamonier
c,
Vishnu K.
Pareek
a,
Moses O.
Tade
a and
Jian
Liu
 *a
*a
aDepartment of Chemical Engineering, Curtin University, Perth, WA 6845, Australia. E-mail: jian.liu@curtin.edu.au
bDepartment of Physics, Curtin University, Perth, WA 6845, Australia
cUniversité de Lille 1, Unité de Catalyse et Chimie du Solide, UMR CNRS 8181, 59652 Villeneuve d'Ascq, France
First published on 3rd March 2015
Abstract
We report the synthesis of CaCO3@C yolk–shell particles with a microporous carbon shell through a selective etching method. The CaCO3@C exhibits an enhanced CO2 adsorption, relative to the porous CaCO3 nanoparticles or the porous carbon shell, with a capacity of 19.30 cm3 STP g−1 (0.86 mmol g−1 or 31.64 cm3 STP cm−3 sorbent) under ambient conditions (23 ± 1 °C and 1 atm CO2).
Yolk–shell nanoparticles are materials with nanoparticle cores inside hollow shells. They are promising functional nanomaterials with various functionalities both on the core and shell, which can have a wide variety of applications such as catalysis, drug/gene delivery, energy storage, biosensors, and Raman scattering (SERS) technologies.1–3 Various yolk–shell nanoparticles (YSNs) with different chemical compositions have been reported such as metal NPs@SiO2, metal oxide@SiO2, metal NPs@C, metal NPs@metal oxide, metal NPs@polymer, SiO2@metal oxide, SiO2@C and polymer@polymer2,4–11 by using different synthetic methods, for example, a soft templating method and selective etching methods. To date, most of the reported YSNs are limited to silica, polymer, carbon and a few metal oxides.2,12–14 To enrich the YSNs library and meet the requirements of practical applications, synthesis of YSNs with a new composition is desirable.
Porous carbons with various morphologies and structures have been widely investigated for CO2 adsorption and separation15 due to their large surface areas, ease of synthesis, low-cost, and high stability.15–19 The synthesis of porous carbon aerogels and xerogels via resorcinol–formaldehyde (RF) resins has been extensively reported.16,20–24 Taking the advantages of yolk–shell structures, such as large void space for accommodating of guest molecules, different functionality of both core and shell, constructing yolk–shell particle with porous carbon shell would be very promising for design of CO2 capture and conversion nanoreactors. Calcium-based materials (calcium oxide, calcium hydroxide, and calcium carbonate) have been proved as excellent sorbents for high temperature CO2 capture,25 however, their adsorption performance quickly declines with multiple reuse due to irreversible particle sintering and agglomeration at high temperatures.26,27
We herein report the first example for the synthesis of CaCO3@C yolk–shell particles. The features offered by these particles are: high surface area and pore volume, basic calcium-based core for affinity of CO2, large void space for CO2 storage, and microporous carbon shell28 for preferential passage of small molecules with respect to larger sized molecules.29 These yolk–shell particles could find application as a catalyst support (e.g., nano-metals) or in drug and gene delivery for biomedical applications,30 or high temperature CO2 capture.
As illustrated in Scheme 1, a four-step synthetic process was employed by using CaCO3 particles as core materials. First, a silica layer was coated around the CaCO3 nanospheres by a Stöber method to obtain CaCO3@SiO2 core–shell particles. Next, the CaCO3@SiO2 was coated with a RF resin via a modified Stöber method28 to produce CaCO3@SiO2@RF core–shell–shell particles. This was followed by a carbonisation process under N2 flow, which converted the RF resin into a microporous carbon shell to produce CaCO3@SiO2@C. Finally, the silica layer was removed with the treatment of hot concentrated NaOH. The detailed experimental procedures are presented in the ESI.†
 | ||
| Scheme 1 Steps involved in the synthesis of CaCO3@C yolk–shell particles. | ||
CaCO3 nanospheres were prepared by the rapid mixing of solutions of CaCl2 and Na2CO3 containing surfactant poly(4-styrenesulfonic acid) sodium salt (PSS). The as-synthesised CaCO3 nanospheres have an average particle size of 450 nm as demonstrated by SEM image (Fig. 1a). The particle size distribution of the CaCO3 spheres (Fig. 1b) further confirms the uniformity of the porous CaCO3 particles. The successful preparation of CaCO3@SiO2 as well as CaCO3@SiO2@C from CaCO3 nanospheres was tracked and confirmed by TEM characterization as shown in Fig. 2a–d. Fig. 3a and b show the SEM and TEM images of the obtained CaCO3@C yolk–shell particles. The presence of a movable core inside a thin carbon shell can be easily identified from the SEM images via the particles with broken shells, exposing the core. The TEM image shows a yolk–shell particle with a porous CaCO3 core and a golf ball like porous carbon shell. The size of the core and hollow space is ca. 240 nm and 310 nm in diameter, respectively, and the carbon shell's thickness is around 10 nm. High angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and energy dispersive X-ray spectroscopy (EDX) of the particles are shown in Fig. 3c–f, which indicates the formation of a carbon shell outside the calcium carbonate core.
 | ||
| Fig. 1 (a) SEM of synthesised CaCO3 nano-spheres and (b) particle size distribution of synthesised CaCO3 nano-spheres. | ||
 | ||
| Fig. 2 TEM images for tracking the steps in synthesis of CaCO3@C: (a) CaCO3 particle, (b) CaCO3@SiO2 particle, (c) CaCO3@SiO2@RF particle and (d) CaCO3@SiO2@C particle. TEOS concentration = 4 mL g−1 CaCO3, RF ratio = 0.5. | ||
 | ||
| Fig. 3 (a) SEM, (b) TEM, (c) HAADF-STEM images, (d–f) EDX elemental mapping of carbon, calcium and oxygen respectively, (g) XRD analysis and (h) N2 adsorption isotherm for the CaCO3@C yolk–shell particles. TEOS concentration = 2 mL g−1 CaCO3, RF ratio = 0.5 and etching time = 3 hours. | ||
XRD pattern of the CaCO3@C yolk–shell particles (Fig. 3g) further confirms that the main composition of the core is CaCO3. A small amount of CaO and CaSiO3 (due to reaction of CaO with un-removed SiO2) is also present. Interestingly, when the CaCO3 precursor nanoparticles were subjected to the same calcination conditions as with the yolk–shell particles, a Ca(OH)2 phase was formed (according to the XRD data, Fig. S1†) due to the loss of CO2 resulting from the continuous supply of fresh N2 in the furnace at 600 °C for a long period (4 hours). This suggests that the nanoporous SiO2 layer prevent the escape of the large CO2 molecules during carbonation of CaCO3@SiO2@RF core–shell–shell particles. As a result, the retainment of CO2 gas within the SiO2 layer led to an expansion of the space between the nanocrystals inside the core, increasing its porosity by creating channels and bridges, compared to the CaCO3 precursor nanoparticles (this can be seen by comparing Fig. 2a and d). Furthermore, some of CaO formed at the edge of the core reacted with SiO2 (that could not be removed by NaOH etching) to form CaSiO3. The porosity of the CaCO3@C yolk–shell particles were measured by nitrogen sorption, which revealed a type IV isotherm indicating their mesoporous structures31 (Fig. 3h). The pore size distribution curve in Fig. S2† shows that the material is highly microporous to mesoporous in nature. The high microporosity of the yolk–shell particles is due to the carbon shell.28 The BET surface area, density and total pore volume of the CaCO3@C YSNs are 381 m2 g−1, 0.12 g cm−3 and 0.61 cm3 g−1, respectively, while those of the CaCO3 precursor nanoparticles are 89 m2 g−1, 0.63 g cm−3 and 0.14 cm3 g−1, respectively (see Fig. S3† for N2 sorption isotherm and pore size distribution of the CaCO3 precursor).
In order to control: (1) the hollow space inside shell and (2) the thickness of shell, the synthesis parameters such as silica precursor concentration and RF ratio were varied, respectively (see Fig. S4–S6 in ESI†). It was found that the general size of the hollow space and shell thickness increased when increasing the silica precursor concentration and the RF ratio, respectively. In addition, by increasing the etching time, as shown in Fig. 4, the hollow space increases and the porous CaCO3 core gets more exposed due to the removal of the silica layer around the core. The ability to control these physical parameters is important in terms of improving the mechanical strength of the particles, and tuning its functionalities.
 | ||
| Fig. 4 Effect of etching time on CaCO3@C particles; (a) t = 1 hour, (b) t = 2 hours, (c) t = 3 hours and (d) t = 4 hours. TEOS concentration = 4 mL g−1 CaCO3, RF ratio = 0.5. | ||
For further optimisation and confirmation of the core compositions, TGA of CaCO3 precursor nanoparticles was performed as shown in Fig. S7,† from which it can be seen that the CaCO3 decomposes to CaO between ∼600 °C and ∼780 °C. As a result, the effect of high temperature recalcination on the CaCO3@C yolk–shell particles was investigated. Fig. S8† shows the XRD patterns for CaCO3@C samples recalcined under N2 for 1 hour at 650 °C, 700 °C and 750 °C, respectively. At 650 °C, a CaO peak starts to appear and becomes more prominent at 700 °C. However, when the temperature is further increased to 750 °C, a CaSiO3 phase forms due to reaction of CaO with remaining SiO2 on the core surface. Hence it is shown that the phase and composition of the core can be changed from CaCO3 to mixture of CaCO3 and CaO, further to CaSiO3 by simply tuning the recalcination temperature of the CaCO3@C yolk–shell particles. The presence of CaO in the core increases its basicity, which can be an attractive characteristic for catalytic applications requiring basic conditions or high temperature CO2 capture.
Developing a low cost and high efficient adsorbent for CO2 capture is highly desirable in order to alleviate the crisis of climate change and greenhouse effect. Fig. 5 shows the CO2 adsorption isotherms for the CaCO3 precursor nanoparticles, Ca(OH)2 (calcined CaCO3 precursor) particles, CaCO3@SiO2@C and CaCO3@C yolk–shell particles. The original CaCO3 shows a relatively low adsorption capacity (6.30 cm3 STP g−1 or 45.00 cm3 STP cm−3 sorbent at 23 ± 1 °C and 1 atm CO2) even at higher CO2 pressures due to weak physisorption of the gas on the particles. The adsorption capacity of CaCO3@SiO2@C is even lower (4.50 cm3 STP g−1 sorbent at 23 ± 1 °C and 1 atm CO2) due to the impervious layer of SiO2 which hindered CO2 penetration towards the CaCO3 core; hence physisorption was mostly achieved on the surface of the carbon shell. The calcined precursor, Ca(OH)2 particles had the lowest CO2 adsorption capacity at 0.40 cm3 STP g−1 sorbent (at 23 ± 1 °C and 1 atm CO2), probably due to the loss in surface area resulting from particles agglomeration at high calcination temperature. However, the improvement in the CO2 uptake is obvious with the yolk–shell particles due to removal of the SiO2 layer for CO2 adsorption. The CaCO3 core of CaCO3@C was more porous than the CaCO3 precursor (as can be seen by comparing Fig. 2a and d). Furthermore, the core–shell architecture allowed each CaCO3 core to be completely surrounded by a CO2 atmosphere for enhanced adsorption. The maximum amount of CO2 adsorbed with CaCO3@C at ambient conditions (23 ± 1 °C and 1 atm CO2) is 19.30 cm3 STP g−1 sorbent (0.86 mmol g−1 or 31.64 cm3 STP cm−3 sorbent). The volumetric capacity of CaCO3 based sorbents is comparable with other adsorbents such as activated carbon,15 much higher than high surface area zeolite 13X (with 5.00 cm3 STP cm−3).32 Fig. S9† shows the determination of the optimum etching time according to CO2 adsorption isotherms. It was found that 3 hours of NaOH etching was enough to expose the CaCO3 core for optimum CO2 adsorption. The CO2 adsorption amount is due to the physisorption, which follows the sequence of CaCO3@C > CaCO3 precursor nanoparticles > CaCO3@SiO2@C > Ca(OH)2 particles, indicating that the combination of CaCO3 core, hollow space and carbon shells is favourable for high CO2 adsorption at low temperature.
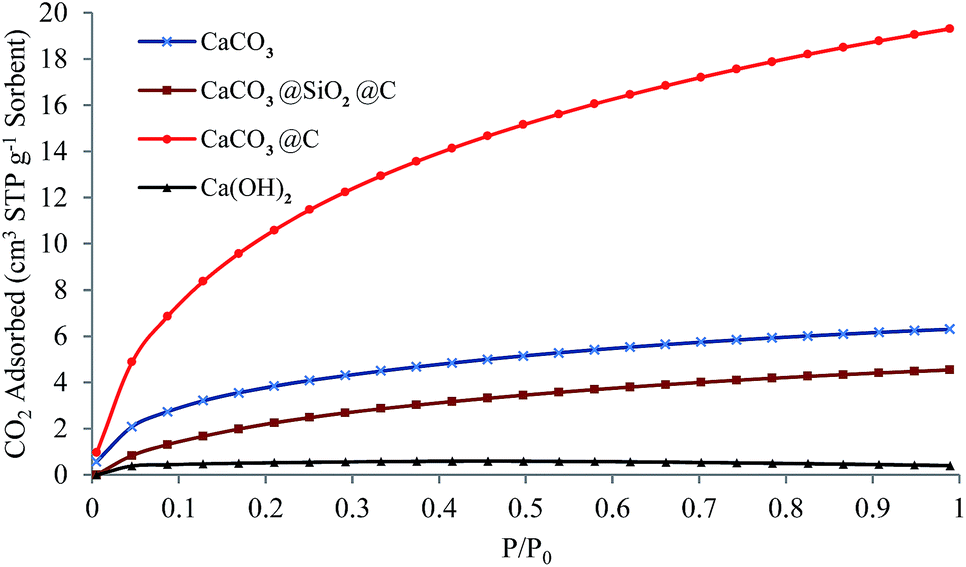 | ||
| Fig. 5 CO2 adsorption isotherms (T = 23 ± 1 °C) for CaCO3 precursor nanoparticles, CaCO3@SiO2@C particles, CaCO3@C yolk–shell particles and Ca(OH)2 particles. For the core–shell–shell and yolk–shell particles, TEOS concentration = 6 mL g−1 CaCO3, RF ratio = 0.5 and etching time = 3 hours. | ||
Conclusions
CaCO3@C yolk–shell particles have been successfully synthesised by a selective etching method. The yolk–shell structures exhibited enhanced CO2 uptake of 19.3 cm3 STP g−1 (0.862 mmol g−1 or 31.64 cm3 STP cm−3 sorbent) at 23 ± 1 °C under 1 atm CO2, compared with 6.3 cm3 STP g−1 for the original CaCO3 core, or 4.5 cm3 STP g−1 for the porous carbon shell. This was due to the relatively high surface area and pore volume, as well as the hollow space of the yolk–shell particle. It was shown that the core composition of the yolk–shell particles can be varied from CaCO3, CaO, or CaSiO3 by simply tuning the recalcination temperature. These yolk–shell particles with porous calcium-based materials core and microporous carbon shell make them potentially attractive materials for environmental remediation (SOx, NOx removal), biomedical applications and nanocatalysis. Further investigation on the high temperature adsorption of CO2, SOx is ongoing.Acknowledgements
The authors acknowledge the facilities, scientific and technical assistance of the Curtin University Electron Microscope Laboratories, a facility partially funded by the University, State and Commonwealth Governments. The authors also wish to acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments. JL gratefully acknowledges the support of France-Australia Science Innovation Collaboration (FASIC) program Early Career Fellowships.Notes and references
- J. Liu, S. Z. Qiao, S. Budi Hartono and G. Q. M. Lu, Angew. Chem., 2010, 122, 5101–5105 CrossRef PubMed.
- J. Liu, S. Z. Qiao, J. S. Chen, X. W. D. Lou, X. Xing and G. Q. M. Lu, Chem. Commun., 2011, 47, 12578–12591 RSC.
- J. Liu, H. Q. Yang, F. Kleitz, Z. G. Chen, T. Yang, E. Strounina, G. Q. M. Lu and S. Z. Qiao, Adv. Funct. Mater., 2012, 22, 591–599 CrossRef CAS PubMed.
- X. Fang, S. Liu, J. Zang, C. Xu, M.-S. Zheng, Q.-F. Dong, D. Sun and N. Zheng, Nanoscale, 2013, 5, 6908–6916 RSC.
- C. Galeano, C. Baldizzone, H. Bongard, B. Spliethoff, C. Weidenthaler, J. C. Meier, K. J. J. Mayrhofer and F. Schüth, Adv. Funct. Mater., 2014, 24, 220–232 CrossRef CAS PubMed.
- Y. J. Hong, M. Y. Son and Y. C. Kang, Adv. Mater., 2013, 25, 2279–2283 CrossRef CAS PubMed.
- J. Wang, W. Li, F. Wang, Y. Xia, A. M. Asiri and D. Zhao, Nanoscale, 2014, 6, 3217–3222 RSC.
- T. Yang, J. Liu, Y. Zheng, M. J. Monteiro and S. Z. Qiao, Chem.–Eur. J., 2013, 19, 6942–6945 CrossRef CAS PubMed.
- T. Yang, R. Zhou, D. Wang, S. P. Jiang, Y. Yamauchi, S. Qiao, M. Monteiro and J. Liu, Chem. Commun., 2015, 51, 2518–2521 RSC.
- R. Liu, F. Qu, Y. Guo, N. Yao and R. D. Priestley, Chem. Commun., 2014, 50, 478–480 RSC.
- R. Liu, Y.-W. Yeh, V. H. Tam, F. Qu, N. Yao and R. D. Priestley, Chem. Commun., 2014, 50, 9056–9059 RSC.
- Y. Chen, H.-R. Chen and J.-L. Shi, Acc. Chem. Res., 2013, 47, 125–137 CrossRef PubMed.
- J. Lee, S. M. Kim and I. S. Lee, Nano Today, 2014, 9, 631–667 CrossRef CAS PubMed.
- M. Priebe and K. M. Fromm, Chem.–Eur. J., 2015, 21, 3854–3874 CrossRef CAS PubMed.
- A. H. Lu and S. Dai, Porous Materials for Carbon Dioxide Capture, Springer, 2014 Search PubMed.
- M. Antonietti, N. Fechler and T.-P. Fellinger, Chem. Mater., 2013, 26, 196–210 CrossRef.
- A.-H. Lu, W.-C. Li, G.-P. Hao, B. Spliethoff, H.-J. Bongard, B. B. Schaack and F. Schüth, Angew. Chem., Int. Ed., 2010, 49, 1615–1618 CrossRef CAS PubMed.
- A.-H. Lu, T. Sun, W.-C. Li, Q. Sun, F. Han, D.-H. Liu and Y. Guo, Angew. Chem., Int. Ed., 2011, 50, 11765–11768 CrossRef CAS PubMed.
- S. Soll, T.-P. Fellinger, X. Wang, Q. Zhao, M. Antonietti and J. Yuan, Small, 2013, 9, 4135–4141 CrossRef CAS PubMed.
- Z.-L. Yu, Z.-Y. Wu, S. Xin, C. Qiao, Z.-Y. Yu, H.-P. Cong and S.-H. Yu, Chem. Mater., 2014, 26, 6915–6918 CrossRef CAS.
- W. Kiciński and A. Dziura, Carbon, 2014, 75, 56–67 CrossRef PubMed.
- D. Wu, C. M. Hui, H. Dong, J. Pietrasik, H. J. Ryu, Z. Li, M. Zhong, H. He, E. K. Kim and M. Jaroniec, Macromolecules, 2011, 44, 5846–5849 CrossRef CAS.
- S. A. Al-Muhtaseb and J. A. Ritter, Adv. Mater., 2003, 15, 101–114 CrossRef CAS PubMed.
- V. G. Pol, L. K. Shrestha and K. Ariga, ACS Appl. Mater. Interfaces, 2014, 6, 10649–10655 CAS.
- Y. Boyjoo, V. K. Pareek and J. Liu, J. Mater. Chem. A, 2014, 2, 14270–14288 CAS.
- A. Samanta, A. Zhao, G. K. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2011, 51, 1438–1463 CrossRef.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 3478–3518 CAS.
- N. Li, Q. Zhang, J. Liu, J. Joo, A. Lee, Y. Gan and Y. Yin, Chem. Commun., 2013, 49, 5135–5137 RSC.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849–1855 CAS.
- Y. Zhao, Z. Luo, M. Li, Q. Qu, X. Ma, S.-H. Yu and Y. Zhao, Angew. Chem., Int. Ed., 2015, 54, 919–922 CrossRef CAS PubMed.
- A. W. Adamson and A. P. Gast, Physical chemistry of surfaces, Wiley, 1997 Search PubMed.
- J. Garcia-Martinez, Nanotechnology for the Energy Challenge, Wiley-VCH, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, SEM and TEM images, CO2 adsorption data. See DOI: 10.1039/c5ra02427g |
| This journal is © The Royal Society of Chemistry 2015 |