
Perfluorobutyl iodide-assisted direct cyanomethylation of azoles and phenols with acetonitrile†
Abstract
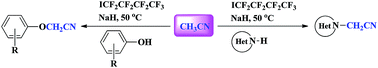
A perfluorobutyl iodide-assisted transition-metal-free cyanomethylation of azoles and phenols with acetonitrile in the presence of NaH has been developed. The reaction proceeded smoothly under mild reaction conditions to give the cyanomethylated products in moderate to high yields. A mechanism involving the cyanomethyl radical through C–H bond cleavage in acetonitrile was proposed.
 Please wait while we load your content...
Please wait while we load your content...

