
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrate uptake using mesoporous silica embedded with zero-valent palladium nanoparticles†
Chee Ling
Tong
a,
Ela
Eroglu
bc,
Xiaofei
Duan
d,
Robert N.
Lamb
d,
Kevin
Jarrett
e,
Craig E.
Buckley
e and
Colin L.
Raston
*a
aFlinders Centre for NanoScale Science and Technology, School of Chemical and Physical Sciences, Flinders University, Bedford Park, SA 5042, Australia. E-mail: colin.raston@flinders.edu.au
bSchool of Chemistry and Biochemistry, The University of Western Australia, Crawley, WA 6009, Australia
cARC Centre of Excellence in Plant Energy Biology, The University of Western Australia, Crawley, WA 6009, Australia
dSurface and Chemical Analysis Network, School of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia
eDepartment of Imaging and Applied Physics, Fuels and Energy Technology Institute, Curtin University, GPO Box U1987, Perth 6845, WA, Australia
First published on 11th February 2015
Abstract
In situ reduction of palladium(II) acetylacetonate during the synthesis of SBA-15 mesoporous silica affords a material impregnated with palladium nanoparticles. The synthesis is at neutral pH and ambient conditions using a continuous flow vortex fluidic device with high shear dynamic thin films, followed by calcination. The SBA-15 is available with considerably reduced processing time relative to the conventional synthesis of mesoporous silica, with the material being effective for nitrate-nitrogen [NO3−-N] removal from aqueous solutions, at 41% within the first 16 hours, or around 36% for the same period after being recycled.
SBA-15 is a mesoporous silica prepared using a non-ionic amphiphilic copolymer template,1 possessing novel physical characteristics and hydrothermal stability. Well defined pores in the material relate to its application in catalysis, separation science, drug delivery, and biosensors.2–4 Advances have been made in improving the synthesis with finer control on the structure and morphology of the material.5–9 Most of the focus has been on the chemistry, as in functionalizing the material,10 or adding swelling agents for pore expansion.11–13 Nonetheless, the synthesis follows the conventional hydrothermal process, which typically takes 48 hours for an individual batch, and reducing this time has been challenging. To this end, we recently made progress in reducing the processing time, using a vortex fluidic device (VFD) for the pre-calcining stage, where the reactions are under shear and are not diffusion controlled.14 Indeed there is limited literature on the synthesis of silica involving such external shear enhancement. Amorphous silica has been prepared using shear flow in a Couette cell under neutral conditions, albeit without much detail provided on the characterization of the material.15 Kim et al. prepared mesoporous silica involving shear flow in a Couette cell operating under laminar flow conditions, although this involved multiple steps and different operating temperatures.16 The aforementioned synthesis of SBA-15 using a VFD under ambient conditions,14 significantly reduced the processing time to ca. 5 hours, prior to the calcination step. Here the turbulent shear condition within the device circumvents the need for hydrothermal aging.14 The simplicity of the VFD processing approach offers scope for customizing the synthesis, depending on the target application, for example nitrate remediation, which is the focus herein.
Heavy usage of artificial fertilizers has resulted in nitrate contamination of ground and surface water.17 The standard regulations for nitrate-nitrogen concentration [NO3−-N] in drinking water is less than 10 mg L−1.18 Nitrate removal has been established for a number of techniques, including ion exchange, biological denitrification,19,20 electro-reduction and electro-coagulation,17 chemical denitrification, reverse osmosis, electrodialysis, and catalytic denitrification.21 Among these commonly used treatment methods, they all have the disadvantage of requiring post-treatment.22 This can be overcome using desorption methods.22 Adsorbents such as activated carbon, sepiolite, and slag have been studied in nitrate removal.23 However, much attention has been devoted to the performance of low cost readily materials such as tree bark, wood charcoal, saw dust, alum sludge, and red mud.24 There are also studies on the catalytic reduction of nitrate using palladium based catalyst and hydrogen gas as the reducing agent, but they have been limited to bench-scale processing. This relates to technical challenges such as catalytic fouling.25 Combining two approaches of utilizing an adsorption method followed by a hydrodeoxygenation reaction would be advantageous. Incorporating palladium in SBA-15 as an adsorbent offers scope for recovery and recycling of the material while avoiding fouling of the catalyst. In the present study we report the in situ synthesis of SBA-15 loaded with zero-valent palladium nanoparticles, and the utility of the material in the removal of nitrate from waste effluent. The reduced palladium nanoparticles provide an active site for catalytic reduction of nitrate via hydrodeoxygenation.25 To our knowledge there are no reports on the use of Pd/SBA-15 as an adsorbent for nitrate removal.
The synthesis herein involves the use of the VFD for the in situ incorporation of metal nanoparticles within the pores of SBA-15 using a Pd(II) precursor. We recently reported a detailed study of the synthesis of SBA-15 using the VFD under acidic and ambient conditions, establishing the ability to control the pore size over a range of operating conditions.14 In a related study we used the VFD to prepare mesoporous silica in the absence of a copolymer, for direct impregnation of particles of an anti-microbial compound into the material, at neutral pH and under ambient conditions, for controlled release at physiological pH.26 These findings form the basis of the present study, with now a focus on gaining access to an adsorbent material for nitrate removal. Here the synthesis is carried out under neutral pH which enables direct deposition of metal oxide, in the presence of a copolymer template.
The synthesis of SBA-15 impregnated with Pd(0) nanoparticles is as follows: 2.5 g of Pluronic® P-123 was dissolved in 50 mL of deionized water at 40–45 °C and 100 mg of palladium(II) acetylacetonate [Pd(acac)2] was added to the resulting micelle solution. The mixture was fed into the VFD, Fig. 1, at a flow rate of 0.5 mL min−1 with the 18.0 mm internal diameter (20.0 mm external diameter) borosilicate glass tube (as a standard NMR tube) spun at 5000 rpm at a tilt angle of 45°. Then 0.375 mL of 0.5 M aqueous sodium fluoride (NaF) was added to the resulting pre-stressed Pd(II)/P-123 mixture, and the solution was fed back into the VFD through one jet-1, with 4.0 mL of tetramethoxysilane (TMOS) simultaneously fed into the VFD through a second jet feed-2. The VFD operating condition were also at 5000 rpm and 45° tilting angle, but now under a H2/N2 atmosphere, with the hydrogen for reducing Pd(II). Detailed synthetic procedures can be found in the ESI.† The overall processing time of the synthesis was 5 hours, with the residence time of a finite volume of liquid in the tube at ca. 3 minutes (Fig. 1). The resulting materials were then calcined at 550 °C under H2 for 6 hours, to remove the polymer template. Initially we used H2PdCl4 in 35% aqua regia as the palladium precursor and synthesize the Pd/SBA-15 using the same synthetic approach under acidic conditions, but the resulting product did not show high mesoregularity, with the palladium particles agglomerated rather than monodispersed inside the channels. In addition, the surface morphology of the material established from scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images suggests incomplete condensation using H2PdCl4 precursor (ESI Fig. S1†).
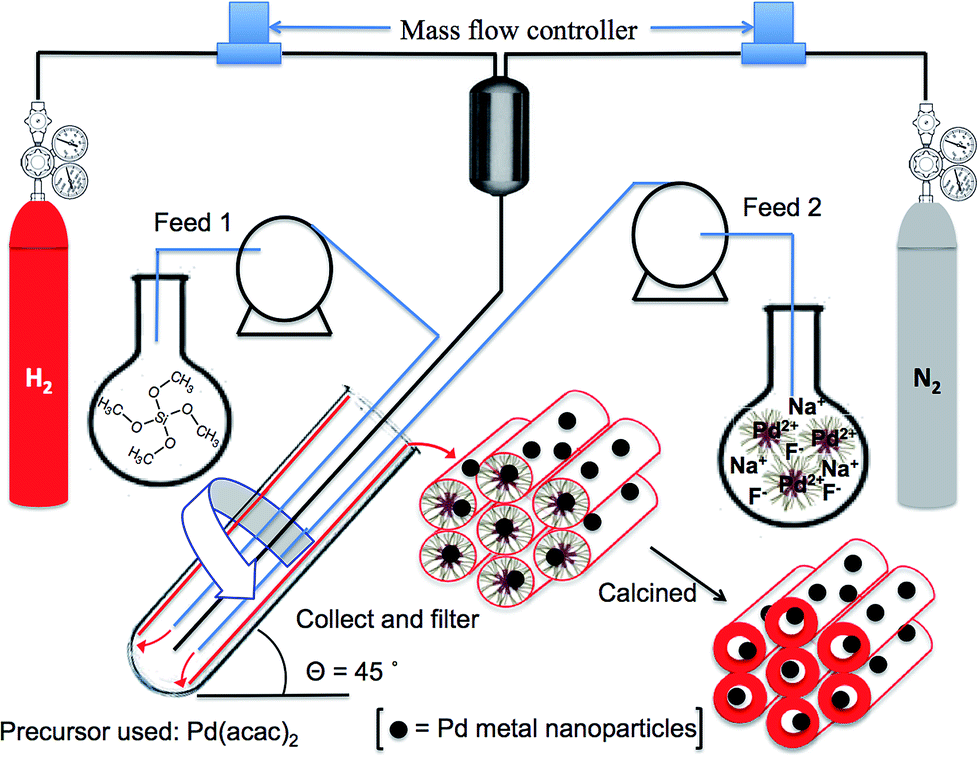 | ||
| Fig. 1 Schematic of the continuous synthesis of Pd/SBA-15 using a vortex fluidic device (VFD). | ||
A small angle X-ray scattering (SAXS) pattern of the calcined material was collected and a strong diffraction peak for the (1 0 0) plane appeared at q = 0.0555 ± 0.0014 Å−1 (Fig. S2†), which corresponds to a d-spacing of 113.2 ± 2.86 Å (Table S1†) and a unit cell parameter, ao of 130.7 ± 3.30 Å (Fig. S3†). No obvious secondary diffraction of (1 1 0) and (2 0 0) plane were observed in the sample. These second order peaks will be washed out by Guinier features (from the size of the Pd particles) causing a decrease in scattering contrast between the pore walls and the pore channels when palladium nanoparticles are incorporated inside the pores.27 This is proven by comparing the SAXS patterns of SBA-15 and Pd/SBA-15 (Fig. S3†).
The surface area, pore volume and pore diameter of the heterogeneous catalyst were investigated using nitrogen adsorption–desorption. The calcined material exhibits a typical type IV isotherm with H1 hysteresis loop according to IUPAC classification (Fig. 2), indicating a uniform mesostructure and cylindrical channels.28 The step increase at relative pressure around 0.45–0.5 was attributed to the capillary condensation of N2 into the mesopores. The material has a Brunauer–Emmett–Teller (BET) surface area of 700.1 m2 g−1 and pore volume of 0.76 cm3 g−1. The narrow pore size distribution of the adsorption branch (inset of Fig. 2) according to Barrett–Joyner–Halenda (BJH) method was centred at 5.4 nm.
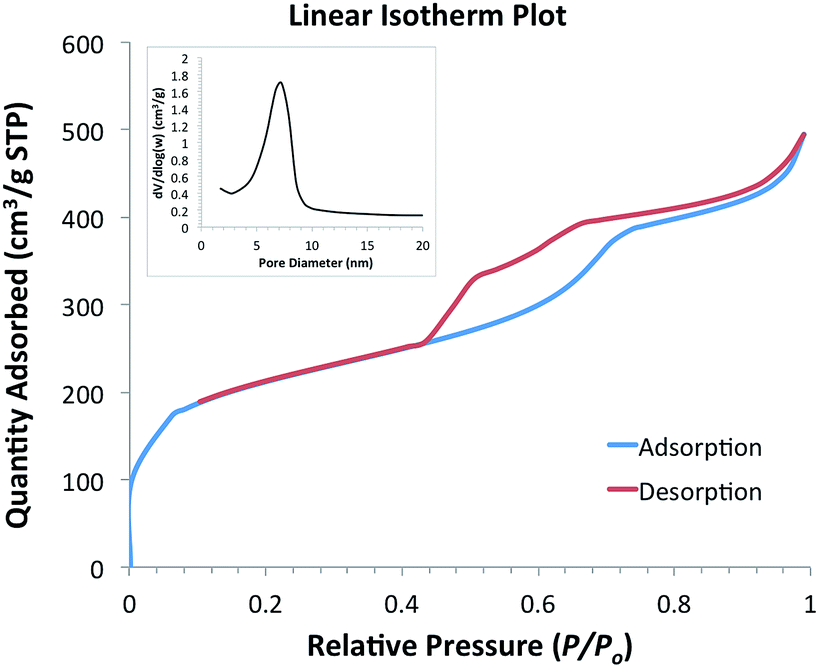 | ||
| Fig. 2 N2 adsorption–desorption of Pd/SBA-15 and the pore size distribution of the adsorption branch (inset). | ||
SEM images showed the formation of short-chain like sections interconnected throughout, Fig. 3(a). Energy-dispersive X-ray spectroscopy (EDS) via SEM was conducted with a full area scanned and mapped for individual elements, silica (Si), oxygen (O) and palladium (Pd), establishing the presence of the elements (Fig. S4†). Inductively coupled plasma mass spectrometry (ICP-MS) gave 1.53 wt% of palladium content in the calcined material, which impart high palladium incorporation, at 81%, into the final product. Transmission electron microscopy (TEM) revealed the inner pore structure of the material, showing a well-ordered mesostructure with estimated pore diameter around 5.5–6 nm which was close to the average pore diameter reported using the BJH method based on N2 adsorption–desorption analysis. Palladium nanoparticles (PdNPs) were revealed as darker contrast regions in the TEM images, with estimated diameters of ∼4–5 nm, and are embedded within the pore channels (Fig. 3(b)). Ruas et al. utilized an alcohol reduction method to prepare a suspension of Pd(0) solution using Pd(acac)2 as the metal precursor, and polyvinylpyrrolidone (PVP) as a stabilizer, with a particle size distribution at 5.8 ± 1.1 nm, which agrees with our finding.29 In addition, using palladium(II) hexafluoroacetylacetonate [Pd(hfac)2] as precursor, the Pd crystallite sizes impregnated into SBA-15 range from 6.0 to 7.9 nm, and the post deposition required 3 to 8 hours under supercritical carbon dioxide (scCO2).30 The size of the PdNPs formed inside the channels is likely to vary with different precursors as well as the choice of solvent system. This is based on their important role in controlling the nucleation and growth of PdNPs.29,31,32
 | ||
| Fig. 3 (a) SEM image of Pd/SBA-15 and (b) TEM image of Pd/SBA-15. | ||
As mentioned above, the synthesis was carried out under H2 to act as a reducing agent in forming elemental palladium nanoparticles. X-ray photoelectron spectroscopy (XPS) was performed to ascertain the oxidation state of the element in the final sample. Survey scans were carried out over the binding energy range 0–1200 eV with 1.0 eV step size and 100 ms dwell time, but no visible Pd peak was observed. Since most of the palladium nanoparticles were clearly incorporated inside the pore channels, an argon ion beam was used to etch the silica surface. A crater area of 3 × 3 mm with approximately 18 nm of the surface layer being sputtered off, was investigated by high resolution scans at analyzer pass energies of 50 eV. The peaks of Pd3d5/2 and Pd3d3/2 binding energy at 335.7 eV and 341.0 eV were observed by deconvolution, indicating an oxidation state of Pd(0) (Fig. S5†).
Fig. 4 shows the amount of NO3−-N concentration of the aqueous media at various time intervals, after being exposed to Pd/SBA-15 samples. Initially, the NO3−-N concentration in the untreated aqueous media was 23 mg L−1. The increase in the amount of adsorbent loading had an affirmative effect on the amount of nitrate-nitrogen adsorbed from the liquid media. All of these three loading capacities achieved their maximal nitrate removal efficiencies after 16 hours, which stabilized thereafter. As shown in Fig. 4, 50 mg of adsorbent yielded the highest nitrate-nitrogen adsorption value of 9.5 mg L−1 (41% removal efficiency of the initial value), which was followed by 8.0 mg L−1 adsorption (35% removal efficiency) by the 25 mg sample, and 6.5 mg L−1 adsorption (28% removal efficiency) by the 10 mg sample. The control experiment with only-silica samples was also investigated using the same amount of adsorbent loadings, with little nitrate removed. This also confirms that the zero valence palladium nanoparticles of the sample are responsible for the nitrate removal. In this context, palladium metal, either monometallic or bimetallic forms, notably Pd–Cu, Pd–Sn or Pd–In, are effective catalysts for the adsorption and catalytic reduction of nitrate.18
 | ||
| Fig. 4 Nitrate-nitrogen [NO3−-N] content of aqueous media in mg L−1versus time for three different Pd/SBA-15 loading concentrations: (1) 10 mg adsorbent (brown column); (2) 25 mg adsorbent (green column); and (3) 50 mg adsorbent (grey column). | ||
From Fig. 4, both 10 mg and 25 mg samples followed a first order adsorption kinetics for the nitrate-nitrogen removal within the first 16th hour, yielding adsorption rates of around 0.4 mg L−1 h−1 NO3−-N (∼1.8 mg L−1 h−1 NO3−) and 0.5 mg L−1 h−1 NO3−-N (∼2.2 mg L−1 h−1 NO3−), respectively. On the other hand, the most concentrated sample (50 mg adsorbent) followed a different adsorption pattern than the other samples, and showed separate removal rates for the time periods of 0–4 h and 8–16 h, resulting in nitrate-nitrogen removal rates of approximately 1.3 mg L−1 h−1 NO3−-N (∼5.8 mg L−1 h−1 NO3−) and 1.1 mg L−1 h−1 NO3−-N (∼4.9 mg L−1 h−1 NO3−), respectively. The kinetic calculations and the accuracy of the removal rates can be improved by measuring more data points within shorter time intervals, which is possible in a larger scale experiment using higher volumes of effluent. While still on the slower side, these nitrate-nitrogen removal rates of the current study are relatively comparable with those reported in the literature on monometallic-palladium containing adsorbents, although it is difficult to make a direct comparison within those studies due to the large variations among the experimental parameters such as the variations in the nitrate removal mechanism, composition and type of the adsorbent, amount of other reducing agents such as catalytic hydrogen, initial concentration and the presence of other ions in the nitrate containing effluent, the scale of the process, and more. As an example, catalytic processes with monometallic palladium reported by Devadas et al. had a nitrate removal rate of ∼4.2 mg L−1 h−1 NO3− with Pd on a support of ceria (CeO2) under the flow of 80% H2 + 20% CO2 gas. They also found that significantly high nitrate removal rate was achieved with the same Pd/CeO2 catalyst under pure hydrogen flow, reaching a value of around 46.5 mg L−1 h−1 NO3−.33
Based on the results shown in Fig. 4, 16 hours was chosen as the optimal time-interval for testing the recycling potential of the adsorbent. Here, used samples were washed with deionized water through hand-vortexing for 1 minute, followed by removal of the supernatant after centrifugation at 9390 × g for 5 minutes. Remaining samples were then mixed again with nitrate-containing fresh media (1.5 mL), while the final nitrate-nitrogen content of the second batch was analyzed after 16 hours of that cycle. Recycling results showed that the Pd/SBA-15 samples were still effective for the removal of nitrate during their second cycle (Table 1), albeit with a slight decrease in their maximum removal efficiencies. The Pd/SBA-15 samples were retrieved and analysed using XPS to investigate if the palladium nanoparticles were oxidized after the treatment. From the spectra, the peaks of Pd3d5/2 and Pd3d3/2 binding energy at 335.5 eV and 340.7 eV were observed by deconvolution, indicating palladium in its elemental form (Fig. S6†).
| [NO3−-N] removal efficiencies (mg L−1) | Amount of Pd/SBA-15 | ||
|---|---|---|---|
| 10 mg | 25 mg | 50 mg | |
| a The numbers given here represent the amount of nitrate-nitrogen [NO3−-N] ions removed from the solution, rather than the amount present in the solution. b Errors reported were deviations from the mean. c Reported overall errors for each batch are calculated using the standard error propagation formula. | |||
| 1st cycle, maximum [NO3−-N] removalb | 6.5 ± 0.6 | 8.0 ± 0.6 | 9.5 ± 0.5 |
| 2nd cycle, maximum [NO3−-N] removalb | 4.5 ± 0.3 | 6.5 ± 0.4 | 8.2 ± 0.4 |
| Total [NO3−-N] removal for two consecutive batchesc | 11.0 ± 0.7 | 14.5 ± 0.7 | 17.7 ± 0.6 |
Conclusions
An efficient method involving the use of the VFD has been developed for preparing mesoporous silica loaded with elemental palladium nanoparticles at neutral pH. This material is effective in wastewater treatment in targeting nitrate-nitrogen removal, showing a maximum nitrate-nitrogen removal efficiency of 41% after 16 hours. Recyclability of the same adsorbent was examined, establishing a 36% maximum nitrate-nitrogen removal efficiency. While this is lower, it nevertheless is promising given that Pd/SBA-15 can be easily separated from the liquid effluent.The novel processing technology of VFD has the advantages of operating under ambient conditions and having a five-fold reduction in processing time relative to the synthesis of conventional hydrothermal processed SBA-15. Also noteworthy is the inherent cost savings in the production of SBA-15 and the analogous material loaded with palladium nanoparticles. This is potentially attractive to industry, as is the ability to generate metal nanoparticles almost exclusively embedded in the pores of the material, rather than on the surface, thereby minimising different outcomes for particles in different environments. This work further highlights the growing applications of the VFD, which also includes its ability to refold protiens.34
Acknowledgements
This work is supported by the Australian Research Council, The Perth Mint, and the Government of South Australia. The authors acknowledge the facility and support from the Australian Microscopy & Microanalysis Research Facility (AMMRF) at Flinders University. C.E.B. acknowledges ARC RIEF grant R00107962 which enabled the SAXS studies to be conducted.Notes and references
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS
.
- J. r. P. Thielemann, F. Girgsdies, R. Schlögl and C. Hess, Beilstein J. Nanotechnol., 2011, 2, 110–118 CrossRef CAS PubMed
- L. Y. Chen, S. Jaenicke and G. K. Chuah, Microporous Mater., 1997, 12, 323–330 CrossRef CAS
- S. Lin, L. Shi, M. M. L. Ribeiro Carrott, P. J. M. Carrott, J. Rocha, M. R. Li and X. D. Zou, Microporous Mesoporous Mater., 2011, 142, 526–534 CrossRef CAS PubMed
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS
- R. Ryoo, C. H. Ko, M. Kruk, V. Antochshuk and M. Jaroniec, J. Phys. Chem. B, 2000, 104, 11465–11471 CrossRef CAS
- D. Zhao, J. Sun, Q. Li and G. D. Stucky, Chem. Mater., 2000, 12, 275–279 CrossRef CAS
- H. I. Lee, J. H. Kim, G. D. Stucky, Y. Shi, C. Pak and J. M. Kim, J. Mater. Chem., 2010, 20, 8483–8487 RSC
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448–2459 CrossRef CAS
- P. Schmidt-Winkel, W. W. Lukens, D. Zhao, P. Yang, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 121, 254–255 CrossRef
- J. S. Lettow, Y. J. Han, P. Schmidt-Winkel, P. Yang, D. Zhao, G. D. Stucky and J. Y. Ying, Langmuir, 2000, 16, 8291–8295 CrossRef CAS
- L. Cao and M. Kruk, Colloids Surf., A, 2010, 357, 91–96 CrossRef CAS PubMed
- C. L. Tong, R. A. Boulos, C. Yu, K. S. Iyer and C. L. Raston, RSC Adv., 2013, 3, 18767–18770 RSC
- S. Patwardhan, N. Mukherjee and S. Clarson, J. Inorg. Organomet. Polym., 2001, 11, 117–121 CrossRef CAS
- W.-J. Kim and S.-M. Yang, Chem. Mater., 2000, 12, 3227–3235 CrossRef CAS
- A. S. Koparal and Ü. B. Öğütveren, J. Hazard. Mater., 2002, 89, 83–94 CrossRef CAS
- Y. Wang, J. Qu, H. Liu and C. Hu, Catal. Today, 2007, 126, 476–482 CrossRef CAS PubMed
- E. Eroglu, V. Agarwal, M. Bradshaw, X. Chen, S. M. Smith, C. L. Raston and K. Swaminathan Iyer, Green Chem., 2012, 14, 2682–2685 RSC
- M. H. Wahid, E. Eroglu, X. Chen, S. M. Smith and C. L. Raston, Green Chem., 2013, 15, 650–655 RSC
- A. Kapoor and T. Viraraghavan, J. Environ. Eng., 1997, 123, 371–380 CrossRef CAS
- A. Bhatnagar and M. Sillanpää, Chem. Eng. J., 2011, 168, 493–504 CrossRef CAS PubMed
- N. Öztürk and T. E. Bektaş, J. Hazard. Mater., 2004, 112, 155–162 CrossRef PubMed
- Y. Cengeloglu, A. Tor, M. Ersoz and G. Arslan, Sep. Purif. Technol., 2006, 51, 374–378 CrossRef CAS PubMed
- B. P. Chaplin, M. Reinhard, W. F. Schneider, C. Schüth, J. R. Shapley, T. J. Strathmann and C. J. Werth, Environ. Sci. Technol., 2012, 46, 3655–3670 CrossRef CAS PubMed
- C. L. Tong, U. H. Stroeher, M. H. Brown and C. L. Raston, RSC Adv., 2015, 5, 7953–7958 RSC
- A. Martin, G. Morales, F. Martinez, R. van Grieken, L. Cao and M. Kruk, J. Mater. Chem., 2010, 20, 8026–8035 RSC
- R. Che, D. Gu, L. Shi and D. Zhao, J. Mater. Chem., 2011, 21, 17371–17381 RSC
- C. P. Ruas, D. K. Fischer and M. A. Gelesky, J. Nanotechnol., 2013, 2013, 6 Search PubMed
- J. Morère, M. J. Tenorio, M. J. Torralvo, C. Pando, J. A. R. Renuncio and A. Cabañas, J. Supercrit. Fluids, 2011, 56, 213–222 CrossRef PubMed
- S. Yang, J. Dong, Z. Yao, C. Shen, X. Shi, Y. Tian, S. Lin and X. Zhang, Sci. Rep., 2014, 4, 4501 Search PubMed
- L. Zhang, L. Wang, Z. Jiang and Z. Xie, Nanoscale Res. Lett., 2012, 7, 312 CrossRef PubMed
- A. Devadas, S. Vasudevan and F. Epron, J. Hazard. Mater., 2011, 185, 1412–1417 CrossRef CAS PubMed
- T. Z. Yuan, C. F. G. Ormonde, S. T. Kudlacek, S. Kunche, J. N. Smith, W. A. Brown, K. M. Pugliese, T. J. Olsen, M. Iftikhar, C. L. Raston and G. A. Weiss, ChemBioChem, 2015, 16, 393–396 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis and characterization methods included. See DOI: 10.1039/c4ra16531d |
| This journal is © The Royal Society of Chemistry 2015 |