
Brønsted acid-catalysed intramolecular ring opening of 2-(aryloxymethyl)-3-aryloxiranes leading to trans-4-arylchroman-3-ols: scope and limitations†
Runjun Devi,
Tapasi Kalita and
Sajal Kumar Das*
Department of Chemical Sciences, Tezpur University, Napaam, Tezpur, Assam, India-784028. E-mail: sajalkd@tezu.ernet.in; sajalkdas@gmail.com; Fax: +91-3712-267005; Fax: +91-3712-267006; Tel: +91-3712-275066
First published on 20th April 2015
Abstract
An operationally simple and metal-free method for the synthesis of a series of trans-4-arylchroman-3-ols via Brønsted acid (TsOH·H2O) catalysed stereoselective intramolecular Friedel–Crafts alkylation of electron-rich arenes by tethered epoxides is developed. The reactions neither need strict anhydrous conditions nor suffer from the use of expensive Lewis/Brønsted acids.
Over the years, stereoselective synthesis of functionalized chroman derivatives has attracted much attention from both academia and the pharmaceutical industry in view of their broad-spectrum biological activities.1 Chroman-3-ol 1 (Fig. 1) constitutes the core skeleton, and is a key synthetic intermediate of a large number of biologically important natural products and medicinally important synthetic molecules-the most familiar examples being flavon-3-ols 2 and 4-arylflavan-3-ols 3 and their derivatives.2
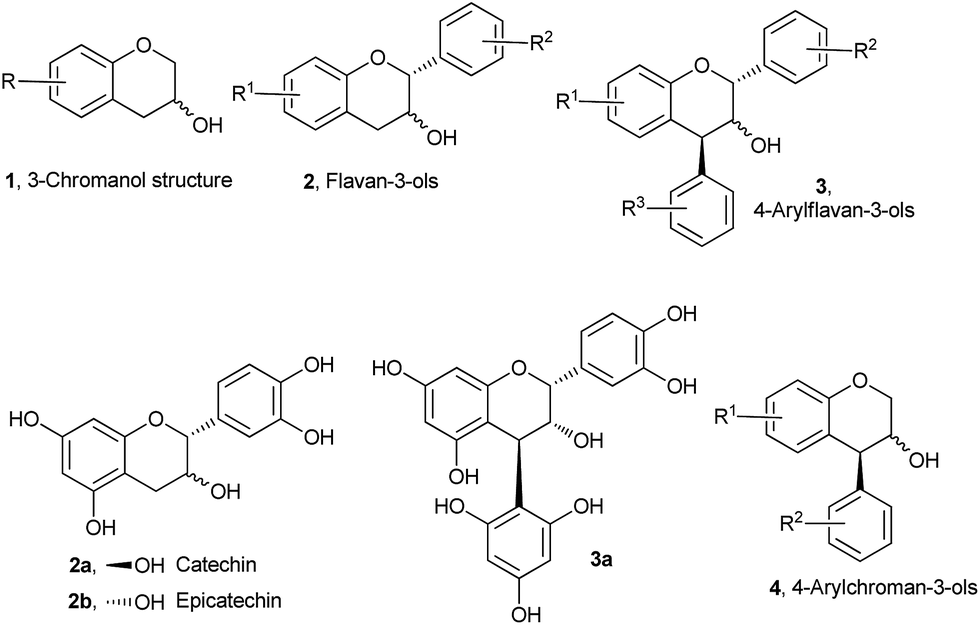 | ||
| Fig. 1 General structures of 3-chromanols, flavon-3-ols, 4-arylflavan-3-ols, 4-arylchroman-3-ols, and representative examples of biologically important natural products of some of these classes of molecules. | ||
One important structural feature of these molecules, as evidenced by their representative members catechin 2a, epicatechin 2b, and compound 3a, is that they all bear highly electron rich aryl rings. These compounds have very widespread biological activities, including antioxidative,3 anticarcinogenic,4 antiarteriosclerotic,5 and antibacterial6 effects. Yet, despite the increasing number of applications for this class of molecules, the systematic synthetic and bio-evaluation studies to access a full array of structural congeners 4-arylchroman-3-ols 4 to maximize properties remain a much less explored area.7 To the best of our knowledge, 4-arylchroman-3-ols, which may be called as “neoflavan-3-ols”, have never been isolated from nature although there has been recent report of isolation of their derivatives.8
On the other hand, due to their ready accessibility in racemic as well as enantiomerically pure forms, and competence to undergo regio- and stereoselective ring-opening reactions, epoxides have found widespread use in organic synthesis.9 Specially, they have been widely used as versatile platforms for the synthesis of natural products and natural product-like molecules via different selective transformations. With the prospective to introduce two adjacent chiral centres with high atom economy, epoxide ring-opening chemistry has been one of the prominent synthetic tools in organic chemistry. Among several possible synthetic transformations of epoxides, Lewis and Brønsted acid-catalysed or promoted intramolecular reactions involving their ring opening are particularly interesting, since they can provide access to a variety of substituted carbo- and heterocyclic compounds. In this context, 2-(aryloxymethyl)-3-aryloxiranes 5 (Scheme 1) have been utilized for the synthesis of racemic and enantiomerically pure trans-4-arylchroman-3-ols (product 6, R2 = aryl, Scheme 1) via transition metal-based Lewis acid-catalysed intramolecular regio- and stereoselective Friedel–Crafts alkylation of electron-rich arenes with epoxides.7b,c On the other hand, there have been reports of intermolecular ring-opening9 and semi-pinacol rearrangement10 of epoxides in the presence of Lewis/Brønsted acids. So there are possibilities of formation of products 7 and 8 from epoxides 5 in the presence of Lewis/Brønsted acids, especially when R2 is not a π donor/bad π donor.
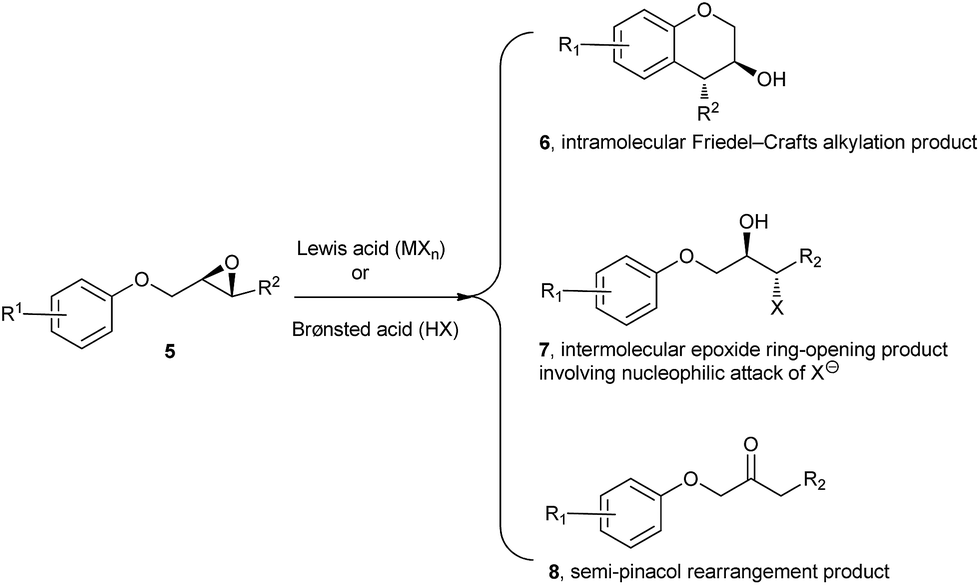 | ||
| Scheme 1 Reaction pathways of aryl glycidyl ethers with Lewis/Brønsted acids. | ||
While the employment of metal-based Lewis acid catalysts offers promises to perform the reaction (product 6, R2 = aryl, Scheme 1) efficiently, it also confines large-scale applications of these reactions and often needs strict anhydrous conditions. We postulated that this issue could be alleviated by the use of easily accessible and affordable Brønsted acids. Furthermore, there is a remarkable lack in the literature concerning the synthesis of trans-4-arylchroman-3-ols via Brønsted acids-catalysed/mediated intramolecular ring opening of epoxides, with the notable exception of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP).7d The use of specialized Brønsted acid like this particular fluorinated alcohol, and its expensive commercial availability can be considered as limiting factors for the widespread use of this method. As a part of one of our research program aimed at synthesising chroman-based polycyclic compounds using epoxide building blocks, we envisioned an operationally simple and metal-free method for the synthesis of a series of trans-4-arylchroman-3-ols via Brønsted acid catalysed stereoselective intramolecular Friedel–Crafts alkylation of electron-rich arenes.
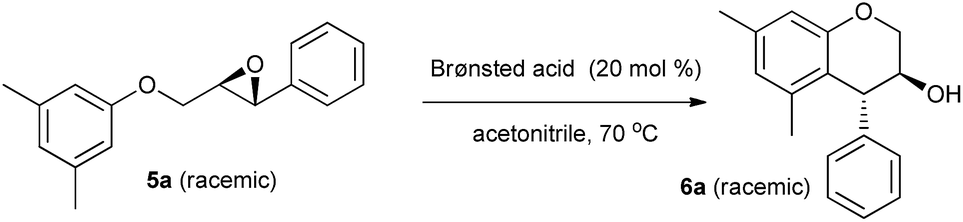
We initiated our study with the optimization of the reaction parameters, including the solvent, the catalyst, reaction temperature and time, and amount of the catalyst used in the reaction. The results are summarized in Tables 1 and 2. First, the intramolecular epoxide ring-opening reaction of racemic 2-((3,5-dimethylphenoxy)methyl)-3-phenyloxirane 5a in acetonitrile was chosen as a model to optimize the reaction conditions (Table 1). As expected, no reaction was observed when a solution of 5a in acetonitrile was heated at 70 °C in the absence of an acid catalyst, resulting only in the quantitative recovery of unreacted 5a (Table 1, entry 1). However, to our delight, we found that the desired product trans-5,7-dimethyl-4-phenylchroman-3-ol 6a was obtained in 80% yield merely by using 20 mol% of p-toluene sulfonic acid monohydrate (TsOH·H2O) as a catalyst (entry 2) within 30 min. With this encouraging result, other easily affordable and accessible Brønsted acid catalysts were further screened. When stronger acids like TFA, TfOH and H2SO4 were used as catalysts, 6a was afforded in lower yields (entries 4–6). In contrast, PhOH and AcOH, the weaker Brønsted acids, as catalysts failed to effect the transformation (entries 7 and 8). Similarly, heterogeneous Brønsted acid catalyst Amberlite IR 120 also produced negative result (entry 9). The results also show that MsOH and TsOH·H2O are equally effective for this reaction (entries 2 and 3). However TsOH·H2O was chosen as a catalyst for subsequent screening of reaction solvents due to its low cost and ease in handling.
|
|||
|---|---|---|---|
| Entry | Brønsted acid catalyst (20 mol%) | Time | Yieldb,c (%) |
| a Reaction conditions: 5a (0.2 mmol), Brønsted acid catalyst, acetonitrile (5 mL), at 70 °C.b Isolated yields after silica gel column chromatography.c For entries 1 and 7–9, we could not detect any product (on TLC) after 30 min and hence the reactions were performed for a longer time. | |||
| 1 | None | 24 h | 0 |
| 2 | TsOH·H2O | 30 min | 80 |
| 3 | MsOH | 30 min | 78 |
| 4 | TFA | 30 min | 65 |
| 5 | TfOH | 30 min | 64 |
| 6 | H2SO4 | 30 min | 60 |
| 7 | AcOH | 5 h | Trace |
| 8 | PhOH | 8 h | 0 |
| 9 | Amberlite IR 120 | 2 h | 0 |
|
||||
|---|---|---|---|---|
| Entry | Solvent | TsOH·H2O (mol%) | Time | Yieldb,c,d (%) |
| a Reaction conditions: 5a (0.2 mmol), TsOH·H2O (X mol%), solvent (5 mL), at 70 °C.b Isolated yields after silica gel column chromatography.c For entries 7–9, we could detect significant amount of unreacted starting material (on TLC) after 30 min and hence the reactions were performed for a longer time.d For entry 6, reaction was completed within 30 min and the yield remained unchanged on running the reaction for longer time (1 h). | ||||
| 1 | CHCl3 | 20 | 30 min | 55 |
| 2 | ClCH2CH2Cl | 20 | 30 min | 66 |
| 3 | 1,4-Dioxane | 20 | 2 h | 0 |
| 4 | THF | 20 | 2 h | 0 |
| 5 | CH3NO2 | 20 | 30 min | 80 |
| 6 | Toluene | 20 | 30 min | 90 |
| 7 | Toluene | 1 | 6 h | 10 |
| 8 | Toluene | 5 | 2 h | 45 |
| 9 | Toluene | 10 | 1 h | 68 |
| 10 | Toluene | 100 | 30 min | 83 |
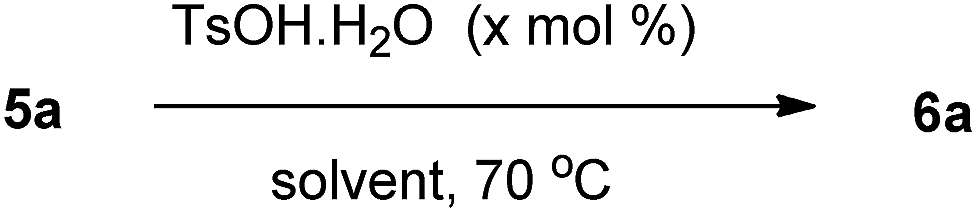
With 20 mol% of TsOH·H2O as catalyst, we screened various solvents (which are commonly used in Friedel–Crafts alkylation) for the intramolecular epoxide ring-opening reaction of racemic 5a leading to 6a (Table 2). While the use of halogenated solvents like CHCl3 and ClCH2CH2Cl gave the lower yield of 6a (entries 1 and 2), cyclic ether solvents like THF and 1,4-dioxane as reaction medium failed to effect the transformation (entries 3 and 4). However, the reaction was higher yielding (80%) in MeNO2 (entry 5). But 6a was obtained in significantly higher yield (90%) when toluene was employed (entry 5). By applying toluene as the reaction medium, further optimizations were then based on the TsOH·H2O loading (Table 2, entries 7–10). A reduced catalyst loading to 1 mol% at 70 °C furnished no product after 30 min and afforded an extremely low yield of 10% of 6a after 6 h (entry 7). The reaction proceeded slowly in the presence of 5 mol% of TsOH, and 45% yield of 6a was obtained after 2 h (entry 8). Treatment of 5a in toluene in the presence of 1.0 equivalent of TsOH·H2O at 70 °C, however, provided 6a in a decreased yield of 83% (entry 10). We are not sure of the exact reason for this decreased yield but it might have arisen from side reactions (such as decomposition, oligomerisation or polymerisation of the starting aryl glycidyl ethers) in the presence of stoichiometric amount of a strong acid, resulting in the formation of uncharacterisable impurities. Whatever may be the reason, these experiments clearly indicate that use of stoichiometric amount of TsOH·H2O is not beneficial for this reaction.
Regarding the influence of temperature on the yield of 6a, we observed that no reaction took place at room temperature (entries 1 and 3, Table 3) with acetonitrile or toluene as solvent. No significant change of yield took place on running the reaction in acetonitrile under reflux conditions (entry 2, Table 3) instead of 70 °C. However, when we conducted the reaction in toluene under reflux condition instead of 70 °C, it resulted in a decrease (from 90% to 80%) in the yield of 6a by generating more side products (entry 4, Table 3).
Consequently, the optimal conditions for the transformation of 5a into 6a supposed the use of TsOH·H2O as catalyst (20 mol%) employing toluene as a reaction medium at 70 °C. It is important to mention that the reaction is regio- and stereocontrolled because only diastreomerically pure trans isomer was isolated. The trans geometry was concluded by comparing the NMR data of 6a with the reported data.7d
Once the best reaction conditions were established, we wanted to study the scope of this method by varying the aryl rings. Toward this purpose, several 2-(aryloxymethyl)-3-aryloxiranes were tested and results are summarized in Scheme 2.
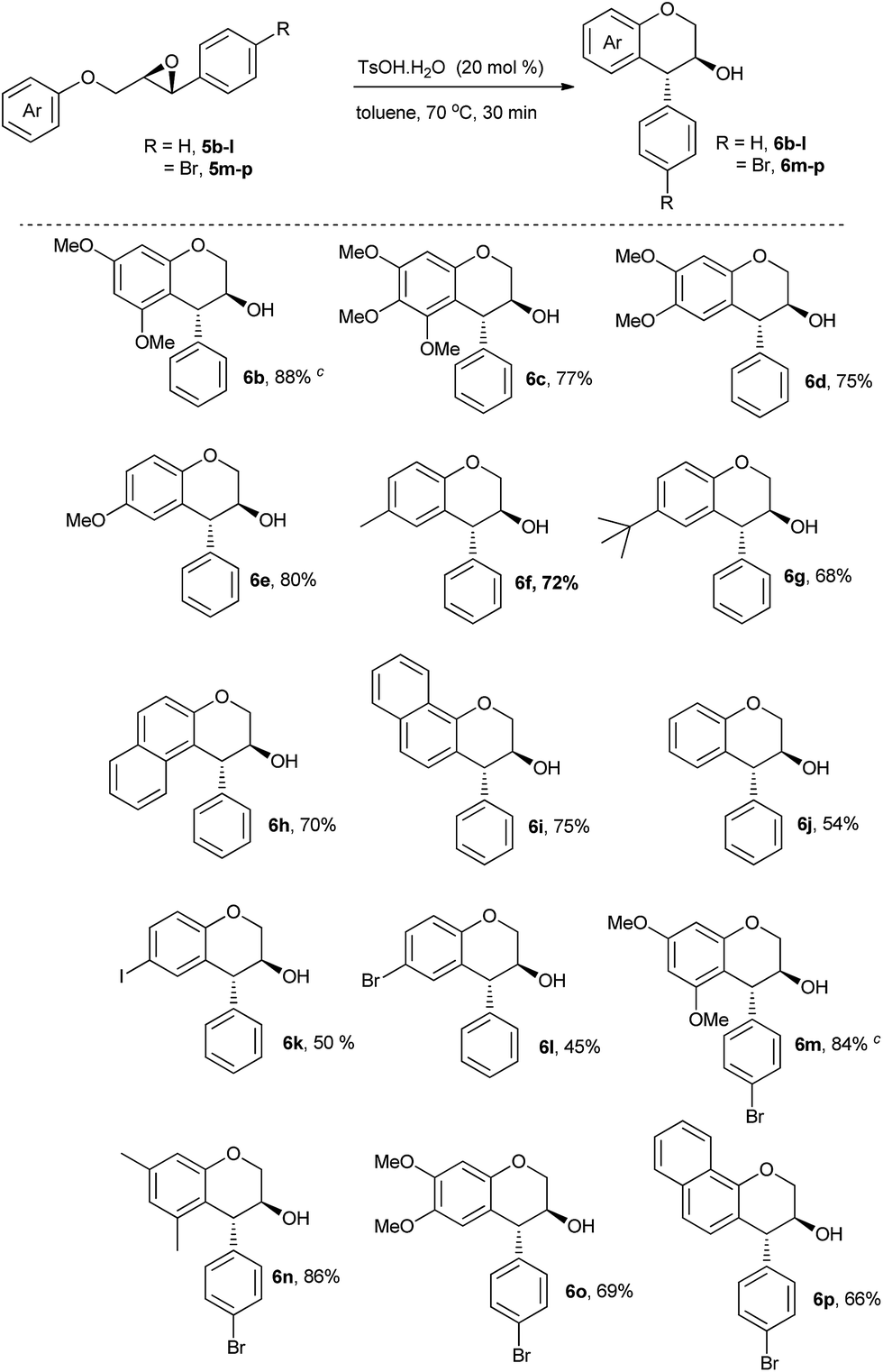 | ||
| Scheme 2 Brønsted acid-catalysed intramolecular ring opening of 2-(aryloxymethyl)-3-aryloxiranes.a,b aUnless otherwise noted, all reactions were conducted by heating a solution of 5b–p (0.2 mmol) and TsOH·H2O (20 mol%) at 70 °C in 5 mL of toluene. bIsolated yields after silica gel column chromatography. cRacemic as well as enantiomerically pure forms were employed. | ||
Because of the presence of higher nucleophilic site on the 3,5-dimethoxyphenyl group, the cyclization of 5b leading to 6b (Scheme 2, product 6b) was high yielding (88%). 3,4,5-Trimethoxyphenyl group also reacted under similar conditions to afford 6c from 5c with a yield of 77% (Scheme 2, 6c). 3,4-Dimethoxyphenyl, 4-methoxyphenyl, 4-methylphenyl, 4-tert-butylphenyl, 2-naphthyl, and 1-naphthyl all were effective nucleophiles to react with the epoxide ring intramolecularly in moderate to good yields (Scheme 2, 6b–l). However, the unsubstituted phenyl group was less reactive as the yield of 6j from 5j was 54% (Scheme 2, 6j). In the reactions with electron-poor 4-bromophenyl, 3-chromanol 6l was obtained only in 45% yield (Scheme 2, 6l). As a result, the reaction with much weaker aryl groups, such as 4-chlorophenyl and 4-fluorophenyl, were not tested. The modest yields with the substrates bearing less nucleophilic aryloxy fragments might be proposed to occur from undesired side-reactions as shown in Scheme 1. Unfortunately, we are not in a position to prove this claim as we could not isolate any side product (despite observing a number of spots on TLC) in pure form and the crude NMR was not conclusive. Nonetheless, the scope of this transformation was further investigated by choosing 2-(4-bromophenyl)-3-(aryloymethyl)oxiranes as substrates (Scheme 2, 5m–p) in which a similar pattern of product formations was observed. To show that chiral oxiranes are compatible with our optimised reaction condition, enantiomerically pure substrates 6b and 6m were also subjected to TsOH·H2O catalysed intramolecular epoxide ring opening reactions which were found to be stereospecific by comparing the products' optical rotation values with the reported data.
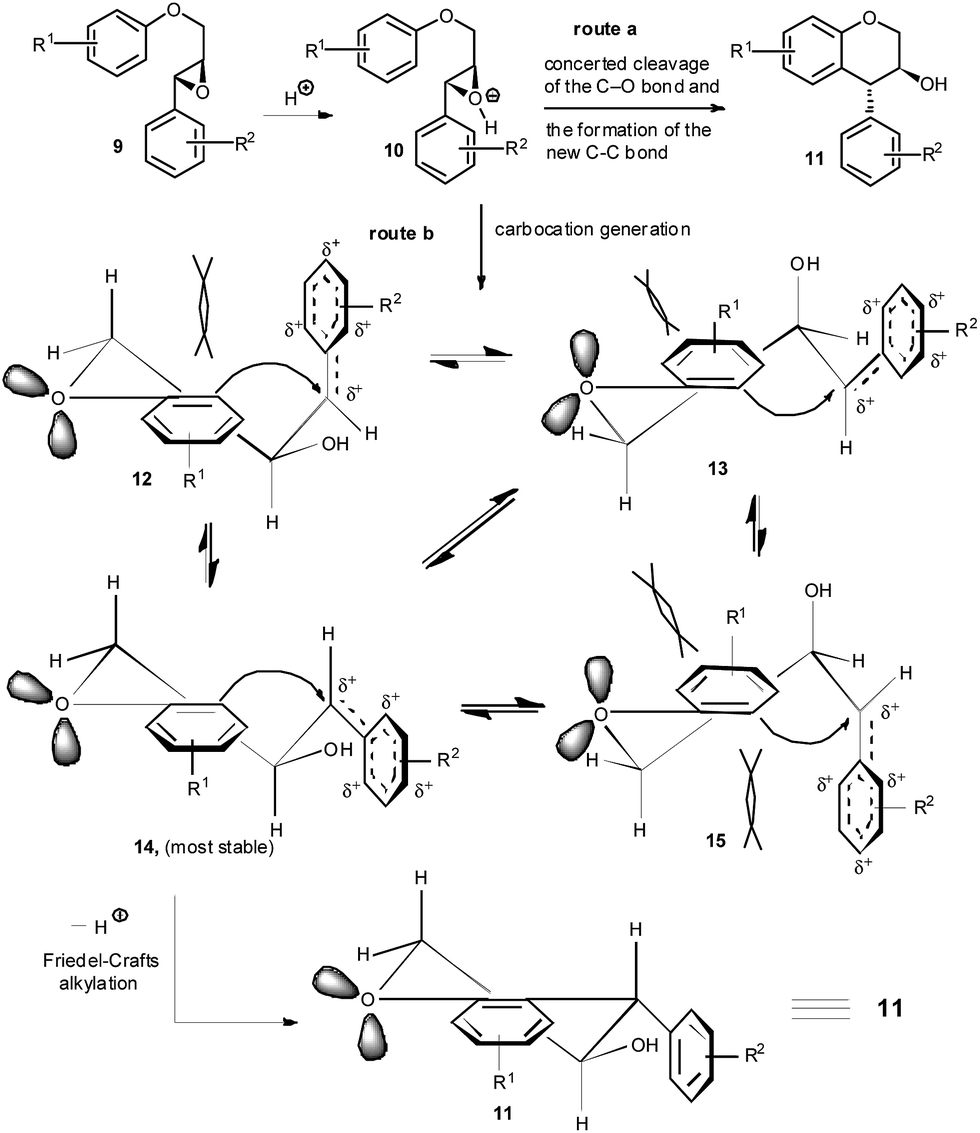 | ||
| Scheme 3 Proposed mechanism for the TsOH-catalysed intramolecular ring opening of 2-(aryloxymethyl)-3-aryloxiranes. | ||
This particular type of ring-opening of epoxides with arenes in the presence of common Brønsted acids has not been previously studied systematically. More importantly, our experiment indicates that use of a weak acid, as suggested by Qu et al.,7d is not compulsory to ensure the cyclisation, in which the arene acts as the nucleophile. Also, activation of epoxides by a strong Brønsted acid like TsOH did not provide the corresponding hydrolysed products. The mechanism for the formation of the trans-4-arylchroman-3-ols via TsOH·H2O catalysed intramolecular Friedel–Crafts alkylation of 2-(aryloxymethyl)-3-aryloxiranes is proposed in Scheme 3.
In case of possible concerted addition of arene to the proton activated oxirane 10 (Scheme 3), as suggested by Qu et al.,7d substrates 9 could furnish observed trans-4-arylchroman-3-ols 11 via 6-endo-tet cyclisation (route a, Scheme 3). While the detailed mechanism awaits further study, we postulated that in the presence of TsOH, the epoxide ring, after protonation, might also be opened to form benzylic carbocation intermediate (route b, Scheme 3). This carbocation can have four conformers (12–15), out of which 14, with all substituents in the pseudoequatorial, may predominate over others.11 Finally, cyclization by a 6-exo-trig mode afforded the observed trans-4-arylchroman-3-ols 11.
To demonstrate the synthetic practicality of this TsOH·H2O catalyzed stereoselective intramolecular Friedel–Crafts alkylation of electron-rich arenes, a gram-scale reaction was carried out by applying 1.5 g of 5a in toluene. In the presence of 10 mol% of TsOH·H2O, this reaction proceeded smoothly at 70 °C to give the desired product 6a in 75% yield (1.1 g) (Scheme 4).
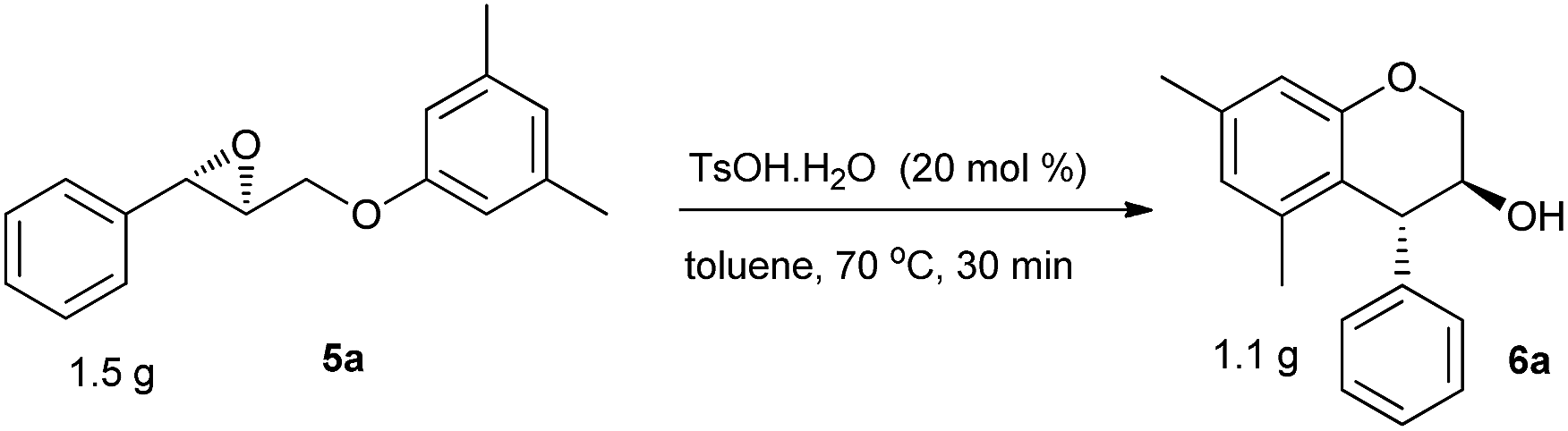 | ||
| Scheme 4 Gram-scale synthesis of racemic 6a. | ||
In view of the above-described interesting results we have already obtained, studies were desired to encompass to glycidyl ethers without any aromatic substituent on the epoxide ring. The question arose if the reaction could still occur without the stimulation of the epoxide ring toward intramolecular ring-opening provided by the electron-rich aryl substituents. Thus, intramolecular ring-opening of glycidyl ether 16a,b were tested by using 20 mol% of TsOH·H2O as catalyst under the already optimized reaction condition (Scheme 5). But to our disappointment, the cyclisation reaction did not proceed even at reflux temperature for 12 h.
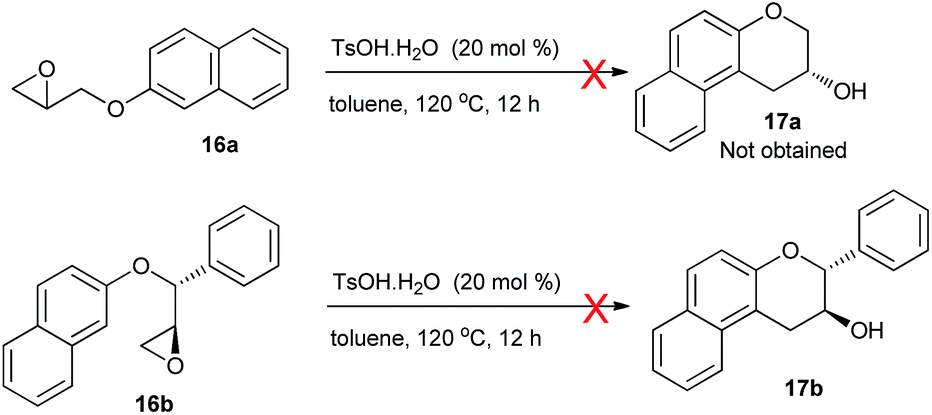 | ||
| Scheme 5 Attempted synthesis of 8a,b. | ||
Bearing in mind the presence of at least two electron-rich phenyl groups in naturally occurring 3-chromanols and their derivatives, the results suggest this method of synthesising trans-4-arylchroman-3-ols containing electron-rich phenyl groups is a fairly good method and provides a useful alternative to the previously reported Lewis acid catalysed protocols. Also, trans-4-arylchroman-3-ols 6m–p hold interesting potential for further elaboration due to the presence of a hydroxyl group on the chroman ring system and a synthetically useful bromo functionality at a remote position. Moreover, all reactions were carried out in open air with AR-grade solvents, which means this transformation is very reliable and easy to operate.
In summary, we have presented a systematic study of the synthesis of a series of trans-4-arylchroman-3-ols via Brønsted acid catalysed stereoselective intramolecular Friedel–Crafts alkylation of electron-rich arenes by tethered epoxides. The results reported here clearly show that the cyclization reactions of 2-(aryloxymethyl)-3-aryloxiranes can be effectively achieved in the presence of 20 mol% TsOH·H2O to get the corresponding trans-4-arylchroman-3-ol derivatives; no diol was formed during the reaction. With this metal-free transformation, a series of trans-4-arylchroman-3-ols were obtained in reasonable to high yields with high regio- and stereoselectivity. The protocol is operationally simple, and neither needs strict anhydrous conditions nor suffers from the use of expensive Lewis/Brønsted acids. Work is in progress aimed at performing mechanistic studies and highlighting applications of this methodology which will be reported in due course as a full paper.
Acknowledgements
Financial support by the Department of Science and Technology [(DST), SB/FT/CS-073/2013], and University Grant Commission [F30-33/2014(BSR)], New Delhi is gratefully acknowledged. SKD thanks Prof. M. K. Chaudhuri, Vice Chancellor, Tezpur University, for providing TU start up grant which helped a lot in setting up our laboratory.Notes and references
- (a) H. C. Shen, Tetrahedron, 2009, 65, 3931 CrossRef CAS PubMed; (b) G. P. Ellis and I. M. Lockhart, The Chemistry of Heterocyclic Compounds, Chromenes, Chromanones, and Chromones, Wiley-VCH, New York, 2007, vol. 31, pp. 1–1196 Search PubMed.
- F. Daneel and D. Slade, Nat. Prod. Rep., 2002, 19, 517 RSC.
- (a) Y. Xu, C.-T. Ho, S. G. Amin, C. Han and F.-L. Chuang, Cancer Res., 1992, 52, 3875 CAS; (b) J. Kinjo, T. Nagao, T. Tanaka, G. Nonaka, M. Okawa, T. Nohara and H. Okabe, Biol. Pharm. Bull., 2002, 25, 1238 CrossRef CAS.
- For selected references, see: (a) D. M. Smith, Z. Wang, A. Kazi, L. H. Li, T. H. Chan and Q. P. Dou, Mol. Med., 2002, 8, 382 CAS; (b) Y. Cao, R. Cao and E. Brakenhielm, J. Nutr. Biochem., 2002, 13, 380 CrossRef CAS; (c) M. Adhami, N. Ahmad and H. Mukhtar, J. Nutr., 2003, 133, 2417S Search PubMed; (d) D. Chen, K. G. Daniel, D. J. Kuhn, A. Kazi, M. Bhuiyan, L. Li, Z. Wang, S. B. Wan, W. H. Lam, T. H. Chan and Q. P. Dou, Front. Biosci., 2004, 9, 2618 CrossRef CAS PubMed; (e) G. Fassina, R. Vene, M. Morini, S. Minghelli, R. Benelli, D. M. Noonan and A. Albini, Clin. Cancer Res., 2004, 10, 4865 CrossRef CAS PubMed.
- For selected references, see: (a) R. Hirano, W. Sasamoto, A. Matsumoto, H. Itakura, O. Igarashi and K. J. Kondo, J. Nutr. Sci. Vitaminol., 2001, 47, 357 CrossRef CAS; (b) J. B. Calixto, M. M. Campos, M. F. Otuki and A. R. Santos, Planta Med., 2004, 70, 93 CrossRef CAS PubMed; (c) K. Y. Chyu, S. M. Babbidge, X. Zhao, R. Dandillaya, A. G. Rietveld, J. Yano, P. Dimayuga, B. Cercek and P. K. Shah, Circulation, 2004, 109, 2448 CrossRef CAS PubMed; (d) Y. Miura, T. Chiba, I. Tomita, H. Koizumi, S. Miura, K. Umegaki, Y. Hara, M. Ikeda and T. Tomita, J. Nutr., 2001, 131, 27 CAS.
- For selected references, (a) Y. Yanagawa, Y. Yamamoto, Y. Hara and T. A. Shimamura, Curr. Microbiol., 2003, 47, 244 CrossRef CAS; (b) S. Gibbons, E. Moser and G. W. Kaatz, Planta Med., 2004, 70, 1240 CrossRef CAS PubMed; (c) T. Taguri, T. Tanaka and I. Kouno, Biol. Pharm. Bull., 2004, 27, 1965 CrossRef CAS; (d) Y. Yoda, Z. Q. Hu, W. H. Zhao and T. Shimamura, J. Infect. Chemother., 2004, 10, 55 CrossRef CAS PubMed; (e) H. Gradisar, P. Pristovsek, A. Plaper and R. Jerala, J. Med. Chem., 2007, 50, 264 CrossRef CAS PubMed.
- (a) D. M. X. Donnelly, J.-P. Finet, P. J. Guiry and K. Nesbitt, Tetrahedron, 2001, 57, 413 CrossRef CAS; (b) Z.-J. Shi and C. He, J. Am. Chem. Soc., 2004, 126, 5964 CrossRef CAS PubMed; (c) R. Marcos, C. Rodrıguez-Escrich, C. Herrerıas and M. A. Pericas, J. Am. Chem. Soc., 2008, 130, 16838 CrossRef CAS PubMed; (d) G.-X. Li and J. Qu, Chem. Commun., 2010, 46, 2653 RSC.
- E. H. Andrianasolo, L. Haramaty, R. Rosario-Passapera, K. Bidle, E. White, C. Vetriani, P. Falkowski and R. Lutz, J. Nat. Prod., 2009, 72, 1216 CrossRef CAS PubMed.
- For selected references, see: (a) A. J. Cresswell, S. G. Davies, J. A. Lee, P. M. Roberts, A. J. Russell, J. E. Thomson and M. J. Tyte, Org. Lett., 2010, 12, 2936 CrossRef CAS PubMed; (b) F. Nikpour, R. Mozafari and B. M. Mogaddam, J. Chin. Chem. Soc., 2009, 56, 404 CAS; (c) F. G. Weber, H. Giese, H. Koeppel, M. Reinhold, R. Strobel, R. Radeglia and W. Storek, J. Prakt. Chem., 1985, 327, 13 Search PubMed; (d) K. Ogawa, S. Ohta and M. Okamoto, Synthesis, 1987, 281 CrossRef CAS.
- For selected references, see: (a) V. Gudla and R. Balamurugan, Chem.–Asian J., 2013, 8, 414 CrossRef CAS PubMed; (b) V. Gudla and R. Balamurugan, Tetrahedron Lett., 2012, 53, 5243 CrossRef CAS PubMed and related references cited therein.
- C. Rondot, P. Retailleau and J. Zhu, Org. Lett., 2007, 9, 247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and copies of 1H and 13C NMR spectra for final compounds. See DOI: 10.1039/c5ra02193f |
| This journal is © The Royal Society of Chemistry 2015 |