
Fabrication of SERS-active conjugated copolymers/gold nanoparticles composite films by interface-directed assembly†
Ya-Guang He,
Sheng-Yu Shi,
Na Liu,
Yuan-Yuan Zhu,
Yun-Sheng Ding,
Jun Yin* and
Zong-Quan Wu*
Key Laboratory of Advanced Functional Materials and Devices, Department of Polymer Material and Engineering, School of Chemistry and Chemical Engineering, Hefei University of Technology, Anhui Province, Hefei 230009, China. E-mail: yinjun@hfut.edu.cn; zqwu@hfut.edu.cn; Fax: +86 551 62902264; Tel: +86 551 62902264
First published on 22nd April 2015
Abstract
We report a new type of functional composite films by taking advantage of the interface-directed assembly between thiol groups functionalized poly(4-isocyano benzoic acid⋯pyridine-4-thiol)-b-poly(3-hexylthiophene) (PPI(–SH)-b-P3HT) conjugated copolymers and gold nanoparticles (Au NPs) at the chloroform/water interface. The PPI(–SH)-b-P3HT copolymers were synthesized through hydrogen bonding induced micellization and subsequent thiol–disulfide exchange reaction. Transmission electron microscopic (TEM) and atomic force microscopy (AFM) observations showed the film was uniform on a large scale and the integrity of surface morphology was not affected by the Au NPs concentration. Interestingly, the film substrate not only exhibited a strongly Au NPs concentration dependent surface-enhanced Raman scattering (SERS) activity but also allowed detection of model molecule, IR-792 perchlorate (IR-792), in the SERS measurement. This proof-of-concept suggests the interfacial assembly route is effective in integrating the properties of organic polymers and inorganic nanoparticles, and for further application.
Introduction
Controlled self-assembly method has been developed for constructing well-defined senior structures from fundamental units arousing significant attention and producing various high performance materials.1–10 Large assembly with multi-dimensional ordered hierarchical architecture and novel collective functions formed by the incorporation of inorganic metal nanoparticles (NPs) into polymer matrices has attracted great scientific and technological interest for bottom-up fabrication of functional materials.11–19Recently, much effort has been devoted to the design and preparation of multi-dimensional structures owing to their potential applications in fabricating practical devices as important building blocks.2,20–22 So far, many successful routes have been developed for the fabrication of senior structures, such as Langmuir–Blodgett deposition,23,24 droplet evaporation,25,26 and interfacial assembly.18,27,28 Inspired by such issues, a facile synthetic method is urgent needed to prepare hybrid nanocomposites, especially ultrathin architecture, in which each component still reserves its inherent properties.
The artificial organization of inorganic metal NPs into diverse ordered senior structures and morphologies, driven by a minimization of interfacial energy at liquid/liquid interface, had been obtained and offered an ideal platform to efficiently induce the self-assembly.11–13,18,29–33 For example, Duan et al. reported a new class of amphiphilic gold nanocrystals with mixed poly(ethylene glycol) and poly(methyl methacrylate) brush coatings, which could spontaneously assemble into 2-D arrays at oil/water interfaces and endow relative nanostructures with tunable morphologies and collective properties.13 Moreover, a series of successful research concerning on polymer and NPs based nanocomposites formed at oil/water interface had also been reported. Brinker et al. reported a general and facile method to prepare free-standing and patternable gold NPs/poly(methyl methacrylate) (PMMA) monolayer arrays via interfacial assembly of NPs in a polymeric photoresist.34 Russell and Emrick et al. developed ultrathin membranes and capsules by ring-opening metathesis polymerization (ROMP) chemistry on norbornene-covered CdSe/ZnS core–shell quantum dots (QDs) to form cross-linked polymer stabilizer at the toluene/water interface.11,35
However, although tremendous successes had been achieved, the challenge still remains to accurately control the assembly of water-dispersible inorganic metal NPs and oil-soluble polymers into uniform multi-dimensional nanocomposites due to the presence of probable local phase separation, aggregation, or precipitation.36,37 Herein, an effective “bottom-up” interface directed assembly of gold NPs (Au NPs) with conjugated polymer matrices is developed to facilely fabricate ordered functional composite film with advantageous surface enhanced Raman scattering (SERS) activity at the chloroform/water interface, as shown in Scheme 1a.
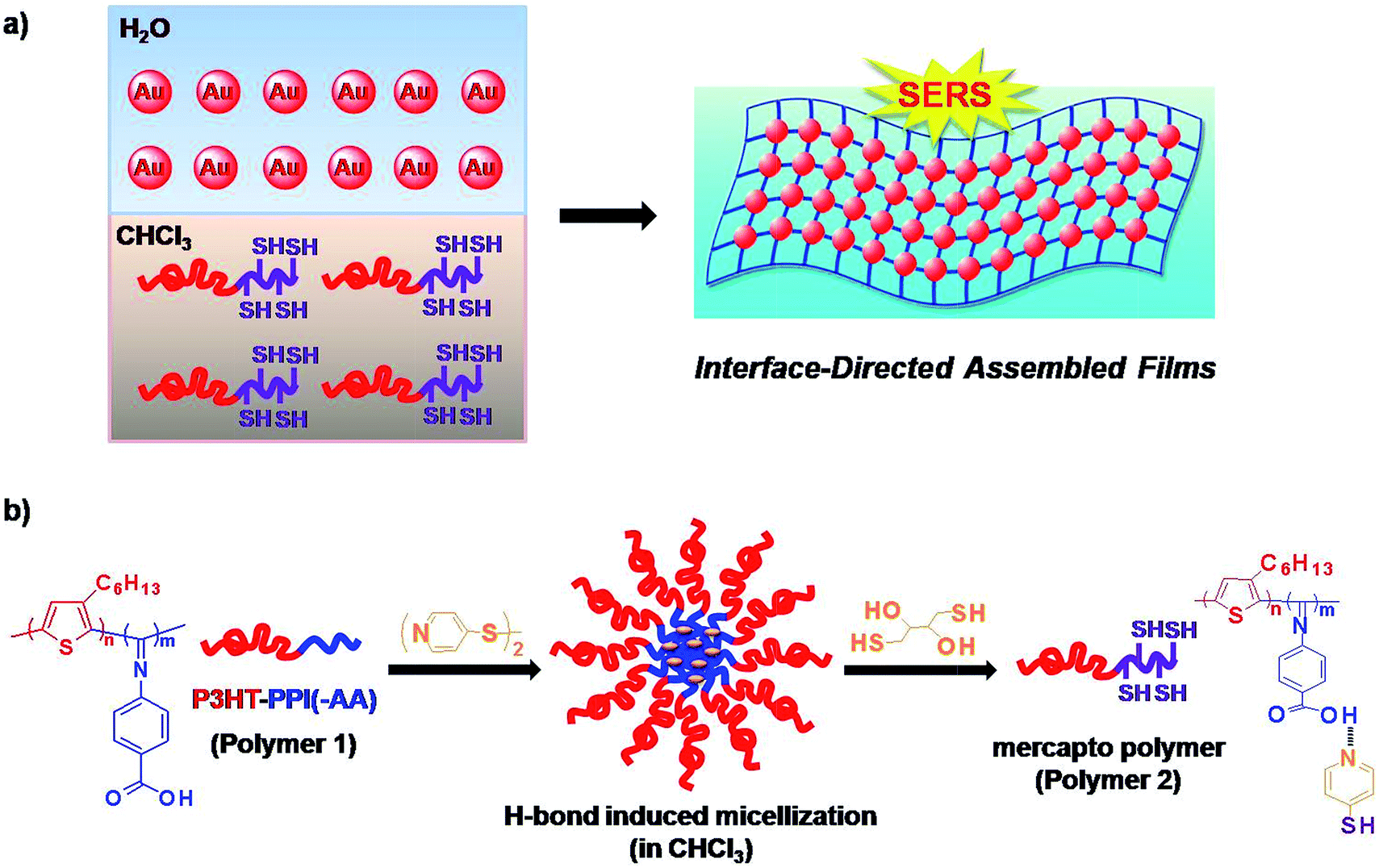 | ||
| Scheme 1 Schematic illustration of (a) interface-directed assembly of SERS-active uniform composite film architecture from PPI(–SH)-b-P3HT conjugated polymers and Au NPs and (b) hydrogen bonding induced micellization mediated fabrication of thiol groups functionalized PPI(–SH)-b-P3HT conjugated polymers. | ||
Conjugated polymers had been reported as a semiconductor that could be used extensively as potential materials for practical device applications.38 Among them, poly(3-hexylthiophene) (P3HT)39–42 and poly(phenyl isocyanide)s (PPIs)43–45 are well-known as fluorescent conjugated polymers and possessing helical structure of the main chain, respectively. Both of them could be self-assembled into various well-defined supramolecular architectures and had been widely studied in theory or used in practical applications.46–48 In this context, a PPI derivatives and P3HT incorporated diblock copolymers, poly(4-isocyano benzoic acid⋯pyridine-4-thiol)-b-poly(3-hexylthiophene) (PPI(–SH)-b-P3HT); polymer 2 in Scheme 1b), were used to react with water-dispersible Au NPs at the chloroform/water interface via the covalent linkage between pendent thiol groups on PPI blocks and Au NPs surface though Au–S bond to form uniform composite film architecture with tunable NPs density dependent SERS activity (Scheme 1a). It had been reported that proton-donating carboxyl groups and proton-accepting pyridyl groups could form stable complexes through 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of carboxyl and pyridyl groups (O⋯N) via hydrogen bonding in a hierarchical fashion.49–54 This is an efficient and rapid pathway to construct supramolecular assembly. With the help of such ponderable idea, the conjugated PPI(–SH)-b-P3HT polymers were prepared from hydrogen bonding interaction between poly(4-isocyano benzoic acid)-b-poly(3-hexylthiophene) (PPI(–AA)-b-P3HT; polymer 1 in Scheme 1b and 4,4′-dipyridyl disulfide (DPDS) in chloroform, and subsequently introduction of dithiothreitol (DTT) to cleave the disulfide bond within the stable complexes, as shown in Scheme 1b. Localized surface plasmon resonance (LSPR) of Au NPs exhibits not only sizable red-shifts upon forming nanoparticle aggregates but also enhanced SERS signals of encoded dyes because of the interparticle plasmonic coupling.55–58 Recently, up to 90-fold of signal enhancement was achieved by the interparticle plasmonic coupling induced by the aggregation of SERS-active dye encoded Au NPs.16 We utilized a modified chloroform/water interfacial adsorption, assembly, and reaction method to fabricate PPI(–SH)-b-P3HT/Au NPs composite films to evaluate the universality and ability of this technique to construct hybrid composites and to prepare functional SERS-active composite films.
:1 ratio of carboxyl and pyridyl groups (O⋯N) via hydrogen bonding in a hierarchical fashion.49–54 This is an efficient and rapid pathway to construct supramolecular assembly. With the help of such ponderable idea, the conjugated PPI(–SH)-b-P3HT polymers were prepared from hydrogen bonding interaction between poly(4-isocyano benzoic acid)-b-poly(3-hexylthiophene) (PPI(–AA)-b-P3HT; polymer 1 in Scheme 1b and 4,4′-dipyridyl disulfide (DPDS) in chloroform, and subsequently introduction of dithiothreitol (DTT) to cleave the disulfide bond within the stable complexes, as shown in Scheme 1b. Localized surface plasmon resonance (LSPR) of Au NPs exhibits not only sizable red-shifts upon forming nanoparticle aggregates but also enhanced SERS signals of encoded dyes because of the interparticle plasmonic coupling.55–58 Recently, up to 90-fold of signal enhancement was achieved by the interparticle plasmonic coupling induced by the aggregation of SERS-active dye encoded Au NPs.16 We utilized a modified chloroform/water interfacial adsorption, assembly, and reaction method to fabricate PPI(–SH)-b-P3HT/Au NPs composite films to evaluate the universality and ability of this technique to construct hybrid composites and to prepare functional SERS-active composite films.
Experiment part
Materials
Chloroauric acid (HAuCl4·4H2O), 4,4′-dipyridyl disulfide (DPDS), and dithiothreitol (DTT) were purchased from Aladdin and used as received. IR-792 perchlorate and sodium citrate was purchased from Sigma-Aldrich and used as received. Chloroform (CHCl3) was distilled over CaH2. Water was deionized with a Milli-Q SP reagent water system (Millipore) to a specific resistivity of 18.0 MΩ cm. Poly(4-isocyano benzoic acid)-b-poly(3-hexylthiophene) (PPI(–AA)-b-P3HT) was reported in our previous literature and the same batch of copolymers with molecule weight (Mn) of 15.7 kDa and polydispersity index (Mw/Mn) of 1.21 were used currently.59Sample preparation
Characterization
Results and discussion
The poly(4-isocyano benzoic acid⋯pyridine-4-thiol)-b-poly(3-hexylthiophene) (PPI(–SH)-b-P3HT) conjugated copolymers were prepared by two steps (Scheme 1b): firstly, simple mixing poly(4-isocyano benzoic acid)-b-poly(3-hexylthiophene) (PPI(–AA)-b-P3HT) with 4,4′-dipyridyl disulfide (DPDS) in chloroform with a 1:1 ratio of carboxyl and pyridyl groups could afford a hydrogen bonding induced micellization. The synthetic procedure applied for PPI(–AA)-b-P3HT copolymers were reported in our previous literature and the same batch of copolymers with molecule weight (Mn) of 15.7 kDa and polydispersity index (Mw/Mn) of 1.21 were used currently.59 Secondly, suitable amount of dithiothreitol (DTT) was added to cleave the disulfide bond within the micellar complex to yield PPI(–SH)-b-P3HT copolymers staying as single polymer chains. The final mixture solution was dialysis against pure chloroform to remove small molecules and stored for further usage. Uniform 22 nm Au NPs were prepared by citrate reduction of HAuCl4 in aqueous solution according to previously reported literatures with little modification,13 the employed synthetic routes was described in ESI† and corresponding evidence for the precise size control and plasmonic property were provided by transmission electron microscopy (TEM), scanning electron microscopy (SEM), dynamic light scattering (DLS), and UV-vis Spectroscopy experiments (Fig. S1 in ESI†), that the Au NPs revealed well-defined spherical structures with average sizes of ∼22 nm.
The hydrogen bonding induced micellization of PPI(–AA)-b-P3HT copolymers was realized by gradual addition of various amount of DPDS against carboxyl groups into chloroform solution, and the corresponding structures obtained were first characterized by dynamic light scattering (DLS) measurement. In Fig. 1a, carboxyl groups functionalized PPI(–AA)-b-P3HT conjugated polymers showed a size distribution of diameter ranging from ∼8 to 17 nm, which could imply the existence of its single chains or possible small multimers formed by self-association. Then, upon the addition of DPDS, the mixed solutions investigated showed distributions at larger diameter ranges with maximum peak value of ∼380 nm, and the peak associated with single polymer chains gradually disappeared with the concentration ratio of [DPDS]/[–COOH] increased from 0 to 2.0. It should be noted here that the single polymers chains are expected to stay separated and no aggregates formed if there are no specific interactions, such as hydrogen bonding, between the polymer chains and DPDS. Thus, it is reasonable to consider that DPDS play a key role in forming such complex. However, a remarkable shift of the distribution to low diameter range was observed once the concentration ratio of [DPDS]/[–COOH] was above 2.0 in the solution, shown in Fig. 1b. This could be attributed to the excess DPDS, which participated in the exchange reaction with existed carboxyl–pyridyl pairs, leading to interrupt the micellar complex, but not served as cross-linkers. Fig. 1c summarized the complex diameter change as a function of concentration ratio of [DPDS]/[–COOH]. Moreover, DLS experiment also revealed that dilution of the complex solution of PPI(–AA)-b-P3HT/DPDS has no effect on the size distribution, this further confirmed that the complexes were very stable in chloroform (data not shown).51
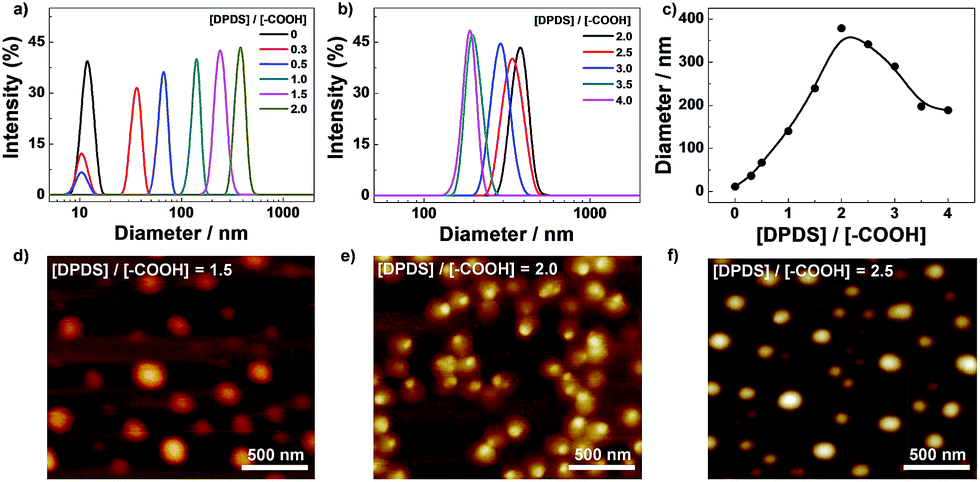 | ||
| Fig. 1 Hydrodynamic diameter distribution of the PPI(–AA)-b-P3HT/DPDS complexes upon addition of various amount of DPDS in CHCl3 solution: (a) [DPDS]/[–COOH] from 0 to 2.0, (b) [DPDS]/[–COOH] from 2.0 to 4.0. (c) Change in relative diameters recorded for PPI(–AA)-b-P3HT/DPDS complexes as a function of [DPDS]/[–COOH]. (d–f) AFM images recorded for PPI(–AA)-b-P3HT/DPDS complexes with different ratios of DPDS to carboxyl groups ([DPDS]/[–COOH] = 1.5, 2.0, and 2.5). The concentration of complex solution is fixed at 0.1 g L−1. | ||
Then, the structural examinations of corresponding complexes were further performed by atomic force microscopy (AFM) measurement. As detailed above, stable complexes could be obtained on the basis of the characteristics of hydrogen bonding between carboxyl and pyridyl groups in chloroform. The typical AFM images obtained from different ratios of DPDS to carboxyl groups ([DPDS]/[–COOH] equal to 1.5, 2.0, and 2.5) were shown in Fig. 1d and f, respectively. These figures indicated the self-assembly of spherical nanoparticles for all these three conditions with maximum diameters of 220 nm, 350 nm, and 300 nm, respectively. These great uniformity nanostructures could be facilely observed and with consistent diameter to that obtained from DLS. It has been well established that AFM determines micelle dimensions in the dry state, whereas DLS reports the intensity-average dimensions of micelles in solution and it contains the contribution of well-solvated coronas. On the basis of chemical intuition, the formed aggregates should consist of PPI(–AA)/DPDS cores and P3HT coronas in chloroform.
According to existing references, the light emission of P3HT-incorporated block copolymer was depending on the self-assembled structures.46–48 However, the absorption and emission spectra of PPI(–AA)-b-P3HT copolymers before and after cross-linking by DPDS in chloroform showed negligible changes with maximum absorption and emission wavelengths appeared at 410 nm and 570 nm (Fig. S2 in ESI†), respectively, and no obviously shift or change upon the gradual addition of DPDS, similar to that of analogous P3HT-incorporated block copolymers in single chains state. The photographs (Fig. S3 in ESI†) also showed a luminous yellow emission under UV illumination at 365 nm and no remarkable changes could be observed even increasing the content of DPDS. The difference in optical properties between the current complexes and previously reported ones should be ascribed to the different location of P3HT blocks, that no induced conformation change of the fully extended P3HT chains happened once acted as the corona of the complex.
The as-formed complexes are based on the disulfide contained DPDS cross-linkers, which is reactive and specific to thiols, thus it can provide a temperate method for generation of thiol groups by thiol–disulfide exchange reactions in the presence of dithiothreitol (DTT).60,61 Addition of a requisite amount of DTT would cause the cleavage of disulfide bond within DPDS moieties to corresponding thiol functionalities and afford targeted PPI(–SH)-b-P3HT copolymers, which will then react with Au NPs at interface to form composite films. To testify this possibility, DTT was gradually added into the PPI(–AA)-b-P3HT/DPDS complex ([DPDS]/[–COOH] = 2/1) solution as against to the concentration of carboxyl groups ([DTT]/[–COOH]) from 0 to 2.0, and the progress of the exchange reaction was also conveniently monitored by DLS, shown in Fig. 2. The results revealed that upon gradual addition of DTT with [DTT]/[–COOH] increased from 0 to 2.0, a remarkable shift of the distribution to low diameter range was observed, accompanied by the recovery of the peaks associated with single polymer chains and then kept a plateau. However, once the [DTT]/[–COOH] was 1/3, the obtained maximum diameter (368 nm) of complex nanostructures was slightly larger than its precursor (350 nm). This can be attributed to the possible swelling of the PPI(–AA)-b-P3HT/DPDS complex in chloroform induced by the partial cleavage of disulfide bond within DPDS moieties but no free PPI(–SH)-b-P3HT copolymer single chains diffusing out. Moreover, further evidence from FT-IR spectra confirmed the existence of mercapto groups within PPI(–SH)-b-P3HT copolymers, and 1H NMR spectra revealed the presence of hydrogen bonding during the self-assembly process due to the chemical shifts of the protons on pyridine moieties (Fig. S4 in ESI†).
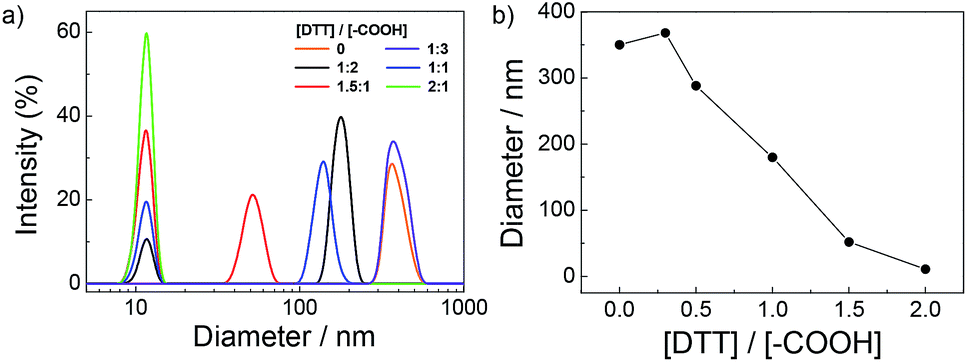 | ||
| Fig. 2 (a) Hydrodynamic diameter distribution of the complexes upon addition of various amount of DTT in CHCl3 solution of PPI(–AA)-b-P3HT/DPDS ([DPDS]/[–COOH] = 2/1). (b) Change in relative diameters recorded for complexes as a function of [DTT]/[–COOH]. | ||
In a mixture of water and chloroform, the Au NPs prefer to stay in water phase due to the favourable solubility of citrate, while the thiol functionalized PPI(–SH)-b-P3HT conjugated copolymers prefer to stay in chloroform phase because of the inherent solubility of organic polymers, as shown in Scheme 1a. The assembly of Au NPs and PPI(–SH)-b-P3HT was spontaneously occurred by taking advantage of the formation of Au–S bond at this biphasic interface driven by a minimization of interfacial energy, leading to a separated composite film. The film showed characteristic golden-color reflected light and purple transmittance due to the strong coupling of Au NPs in close proximity, which accompanied by the color disappeared in the water phase from light red to almost colorless.
In order to verify the existence and distribution of Au NPs in the film, TEM was employed to examine the assembled structures precisely, shown in Fig. 3a–c. The obtained images on a carbon coated copper grid showed the formation of clear film-like structures encapsulated both of Au NPs and polymers distinctly with sizes of up to several micrometers. The local part with much deeper dark color should be ascribed to the wrinkle of polymer film, which is always not visible because of its poor contrast. The films were then investigated by tapping-mode AFM, shown in Fig. 3d. The morphology analysis indicated that the films were very flat and uniform, and the height measurement suggested the presence of “mountains” up to as high as ∼24 nm. This agreed very well with the combining diameters of the naked Au NPs (∼22 nm) and a conjugated polymer layer, strongly suggesting the formation of thin layer sheets. Compared with traditional Langmuir–Blodgett and layer-by-layer deposition, assembly at the liquid/liquid interface requires fewer steps and affords thin membranes.59
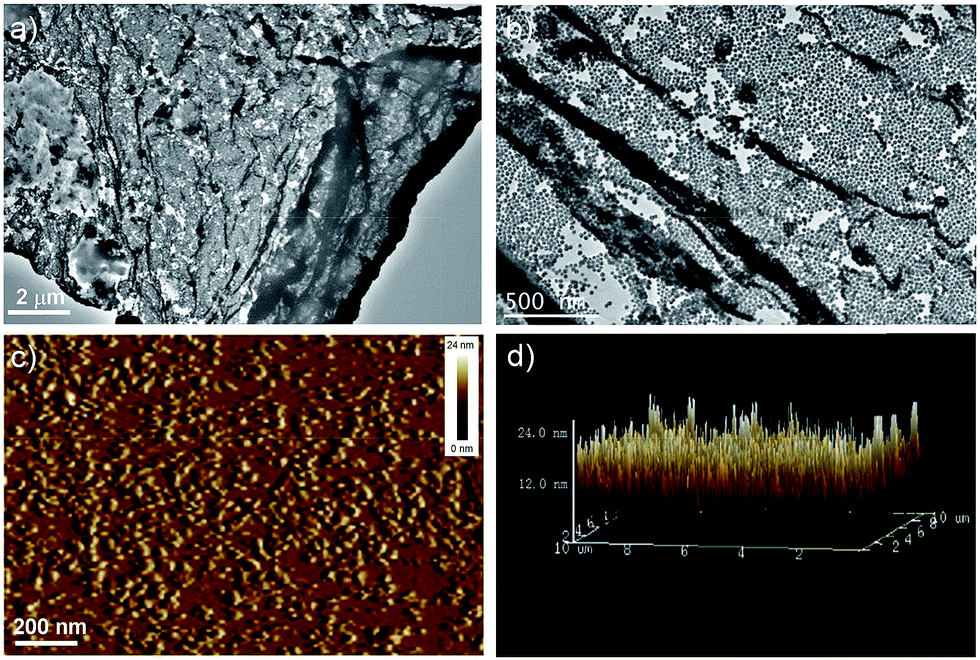 | ||
| Fig. 3 (a and b) TEM and (c and d) AFM images obtained for PPI(–SH)-b-P3HT/Au NPs hybrid films. The concentration of Au NPs aqueous dispersion and PPI(–SH)-b-P3HT solution is 10 nM and 0.1 g L−1, respectively. | ||
On the other hand, control experiments were performed to establish the relationship between the role of PPI(–SH)-b-P3HT and Au NPs and the formation of composite films, in which core cross-linked PPI(–AA)-b-P3HT/DPDS complexes were used to couple Au NPs under the same conditions. Significantly, no assembled nanocomposites could be achieved at the biphase interface, this difference should be due to the shielding effect on the disulfide bond within the complex interior. Therefore, a possible mechanism for the assembly of composite films could be proposed that the pendent thiol groups on PPI(–SH)-b-P3HT chains in the chloroform phase oriented toward the biphase interface and then reacted with Au NPs surface, the synergetic continuous collapse of P3HT blocks once the surrounding environment changed from a good solvent to a poor solvent near the interface to afford the composite films. The obviously transparent feature suggested that they should be extremely thin, probably several layers because of the quite unstable character under the condition used for the microscopy experiments.
An universal phenomenon of assembled Au NPs is that of exhibiting dramatic sizable red shifts upon forming nanoparticle aggregates due to strong interparticle plasmonic coupling.57,58 The optical properties of PPI(–SH)-b-P3HT copolymers in chloroform solution, as polymer film, and as thin film embedded with Au NPs were investigated by UV-vis and relevant results were shown in Fig. 4a. We can see that the absorption maximum of the copolymer film (blue line) exhibit obvious red shift compared to that of polymer solution (red line), this result is due to the strong intermolecular π–π stacking of thiophene rings after the diffusion of solvent molecules out of the assembly. Moreover, the maximum absorption peak showed an obvious red-shift and became wider with the absorption edge (λedge) extended to about 900 nm once embedded with Au NPs as films (black line). The significant red-shift of the SPR band here should be a result of the continuous approach of neighboring Au NPs in the solid state, which reduced the separating distance of adjacent Au NPs and also increased the local refractive indexes. Fig. 4b showed that the PPI(–SH)-b-P3HT copolymers exhibited a strong emission peaked at 570 nm, however, the PL spectrum of the films was quenched after being either polymer film or embedded with Au NPs. This is due to the assembly induced quenching site enhancement46 and fluorescence resonance energy transfer between PPI(–SH)-b-P3HT and Au NPs, respectively.
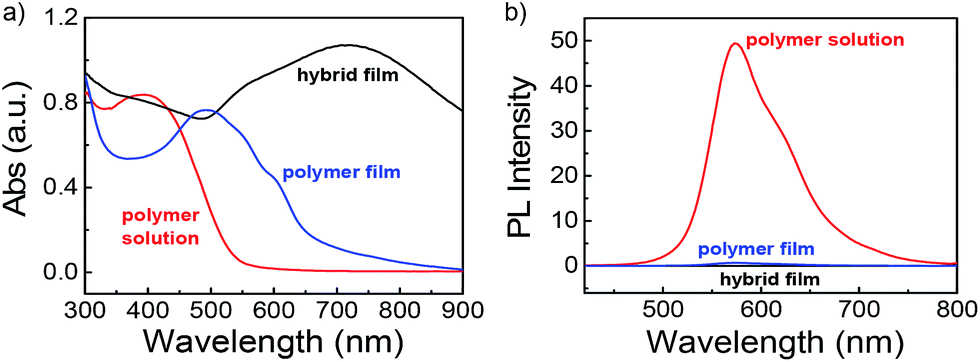 | ||
| Fig. 4 (a) UV-vis and (b) emission spectra recorded for PPI(–SH)-b-P3HT copolymers in CHCl3, as polymer film, and after embedded with Au NPs as composite film. | ||
SERS technique has emerged as a precise and powerful tool with outstanding sensitivity for practical applications in a number of chemical and biological fields.16,57,62,63 Accumulating evidence have demonstrated that the extent of SERS signal amplification is closely correlated to the size as well as the average number of the nanoparticles on a platform.57,58 Thus, the development of novel SERS-active platform is extraordinary significant. Aside from LSPR property, enhanced SERS signals of dyes encoded Au NPs can be detected upon forming aggregates because of interparticle plasmonic coupling. Herein, the effects of Au NPs concentration on the morphology of films formed at the chloroform/water interface as well as their corresponding SERS intensities were investigated. Fig. 5a showed two typical AFM images of the surface morphology of films formed with a polymer concentration of 0.1 g L−1 and Au NPs aqueous dispersions with various concentrations of 10 nM and 3 nM, respectively. According to the molar feed ratio of PPI(–SH)-b-P3HT and Au NPs, AFM revealed different densities of Au NPs embedded on the films. When an Au NPs aqueous dispersion with a higher concentration of 10 nM was used, a film composed of a large amount of Au NPs was observed (top image in Fig. 5a), whose NPs density is much larger than that formed using 3 nM Au NPs aqueous dispersion (bottom image in Fig. 5a). Moreover, as clearly seen from both of the AFM images, the surface morphology of film was not affected by the Au NPs concentration and the amount of Au NPs could be easily tuned using this approach.
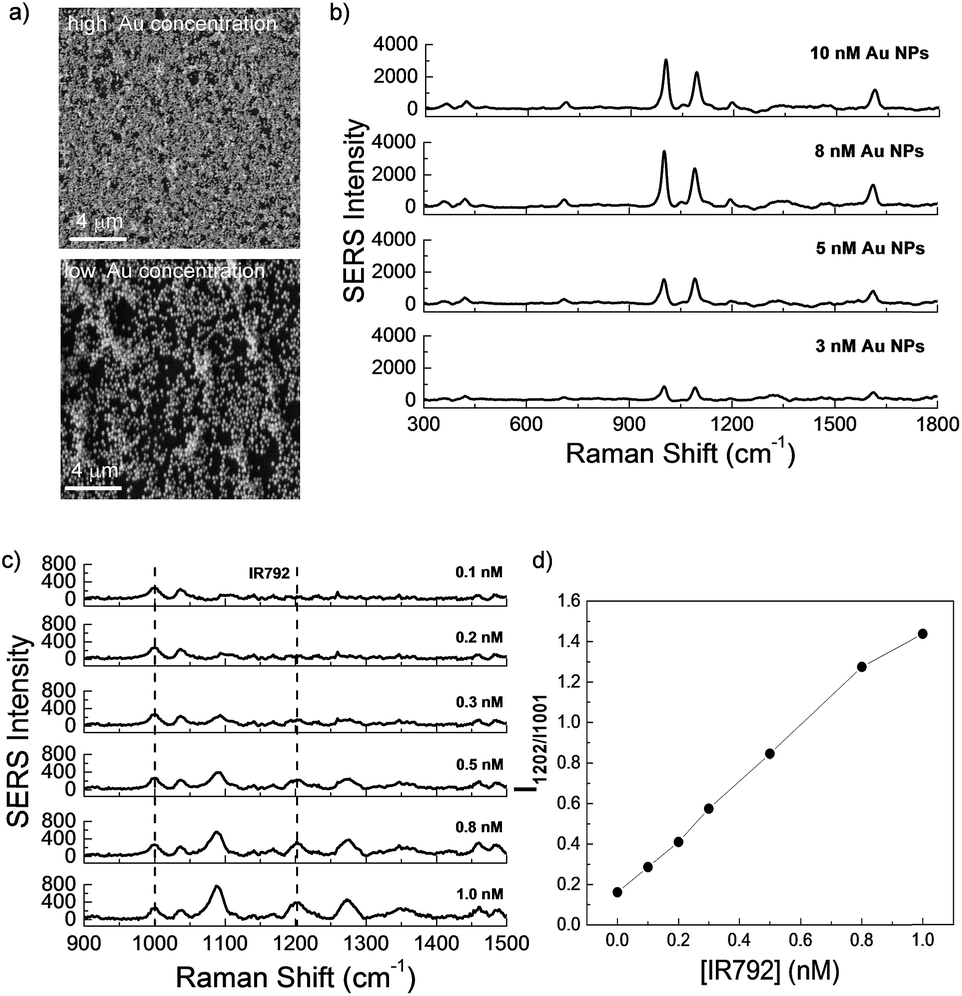 | ||
| Fig. 5 (a) Typical SEM images of the surface morphology of films formed with Au NPs aqueous dispersions with various concentrations of 10 nM (top) and 3 nM (bottom). (b) SERS spectra recorded for SERS-active film embedded with various concentrations of Au NPs. (c) SERS spectra and (d) change in relative SERS intensity recorded for SERS-active film (embedded with 3 nM Au NPs) immersed in different concentration of IR-792 solutions. The concentration of polymer solution is fixed at 0.1 g L−1. | ||
Correspondingly, the SERS signals of these two types of films were detected. 785 nm excitation is selected for current signal detection because 785 nm laser excitations are favorable for nanoparticles aggregates, and the matching between the absorption band and the laser excitation wavelength leads to a resonance with an additional enhancement factor.64 In Fig. 5b, obvious SERS signal intensity from pyridine groups in close proximity to Au NPs surface was presented for films formed with 3 nM Au NPs aqueous dispersion, when the Au NPs concentration was increased to 10 nM, the SERS peak at 1001 cm−1 was amplified ∼4-fold enhancement, which could be explained by an enhancement of the intensity of local field when the gap between the Au NPs decreased.65
To assess the Raman detection and SERS sensitivity of the composite film, SERS spectra of corresponding films immersed in different concentrations of IR-792 perchlorate (IR-792) were measured. As shown in Fig. 5c, the Raman characteristic bands of IR-792 at 1202 cm−1 were clearly presented with the concentration of IR-792 increased, and the correlation ratio (I1202 cm−1/I1001 cm−1) change could be calculated by the presence of pyridine peak as internal standard. Excellent sensitivity could be obtained even when the concentration of IR-792 was reduced to 0.1 nM (Fig. 5d).
Conclusion
A composite film composed of conjugated polymers and metal NPs was established at the planar liquid/liquid interface between a chloroform solution of thiol functionalized PPI(–SH)-b-P3HT and Au NPs aqueous dispersion at ambient temperature. The resulting composite films had a highly ordered architecture with tunable Au NPs density, depending on the initial concentration of Au NPs, and exhibited concentration-dependent surface enhanced Raman scattering (SERS) activity. Undoubtedly, this interfacial assembly strategy offers a versatile route to incorporate inorganic nanoparticles into conjugated polymer matrices, forming nanomaterials potentially useful in relative applications. In addition, this facile approach can be easily applied to various metal NPs or quantum dots (QDs) to develop highly ordered functional films.Acknowledgements
This work is supported in part by National Natural Scientific Foundation of China (21104015, 21172050, 21302035, 21371043, 51303044, and 21304027), Natural Scientific Foundation of Anhui Province (1208085QB29 and 1408085QE80). Z.W. thanks the Thousand Young Talents Program of China for Financial Support. J.Y. expresses his thanks for Specialized Research Fund for the Doctoral Program of Higher Education (20130111120013) and Research Foundation for Returned Overseas Chinese Scholars of the Ministry of Education of China.Notes and references
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- L. M. Xu, L. X. Jiang, M. Drechsler, Y. Sun, Z. R. Liu, J. B. Huang, B. Z. Tang, Z. B. Li, M. A. C. Stuart and Y. Yan, J. Am. Chem. Soc., 2014, 136, 1942–1947 CrossRef CAS PubMed.
- Y. J. Min, M. Akbulut, K. Kristiansen, Y. Golan and J. Israelachvili, Nat. Mater., 2008, 7, 527–538 CrossRef CAS PubMed.
- X. R. Wang, G. H. Liu, J. M. Hu, G. Y. Zhang and S. Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 3138–3142 CrossRef CAS PubMed.
- J. M. Hu, T. Wu, G. Y. Zhang and S. Y. Liu, J. Am. Chem. Soc., 2012, 134, 7624–7627 CrossRef CAS PubMed.
- C. Boyer, M. R. Whittaker, V. Bulmus, J. Q. Liu and T. P. Davis, NPG Asia Mater., 2010, 2, 23–30 CrossRef.
- Y. Mai and A. Eisenberg, J. Am. Chem. Soc., 2010, 132, 10078–10084 CrossRef CAS PubMed.
- S. Park, J. H. Lim, S. W. Chung and C. A. Mirkin, Science, 2004, 303, 348–351 CrossRef CAS PubMed.
- R. Shenhar, T. B. Norsten and V. M. Rotello, Adv. Mater., 2005, 17, 657–669 CrossRef CAS PubMed.
- Y. Lin, A. Boker, J. B. He, K. Sill, H. Q. Xiang, C. Abetz, X. F. Li, J. Wang, T. Emrick, S. Long, Q. Wang, A. Balazs and T. P. Russell, Nature, 2005, 434, 55–59 CrossRef CAS PubMed.
- H. Skaff, Y. Lin, R. Tangirala, K. Breitenkamp, A. Boker, T. P. Russell and T. Emrick, Adv. Mater., 2005, 17, 2082–2084 CrossRef CAS PubMed.
- M. Liu, Q. Wang, Y. Y. Geng, C. W. Wang, Y. I. Lee, J. C. Hao and H. G. Liu, Langmuir, 2014, 30, 10503–10512 CrossRef CAS PubMed.
- L. Cheng, A. P. Liu, S. Peng and H. W. Duan, Acs Nano, 2010, 4, 6098–6104 CrossRef CAS PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- K. H. Ku, J. M. Shin, M. P. Kim, C. H. Lee, M. K. Seo, G. R. Yi, S. G. Jang and B. J. Kim, J. Am. Chem. Soc., 2014, 136, 9982–9989 CrossRef CAS PubMed.
- J. Yin, T. Wu, J. B. Song, Q. Zhang, S. Y. Liu, R. Xu and H. W. Duan, Chem. Mater., 2011, 23, 4756–4764 CrossRef CAS.
- Y. J. Liu, Y. C. Li, J. He, K. J. Duelge, Z. Y. Lu and Z. H. Nie, J. Am. Chem. Soc., 2014, 136, 2602–2610 CrossRef CAS PubMed.
- S. Y. Yang, C. F. Wang and S. Chen, J. Am. Chem. Soc., 2011, 133, 8412–8415 CrossRef CAS PubMed.
- M. Kanahara, M. Shimomura and H. Yabu, Soft Matter, 2014, 10, 275–280 RSC.
- T. Y. Zhou, F. Lin, Z. T. Li and X. Zhao, Macromolecules, 2013, 46, 7745–7752 CrossRef CAS.
- S. Srivastava and N. A. Kotov, Soft Matter, 2009, 5, 1146–1156 RSC.
- M. P. Pileni, Y. Lalatonne, D. Ingert, I. Lisiecki and A. Courty, Faraday Discuss., 2004, 125, 251–264 RSC.
- D. G. Schultz, X. M. Lin, D. X. Li, J. Gebhardt, M. Meron, P. J. Viccaro and B. H. Lin, J. Phys. Chem. B, 2006, 110, 24522–24529 CrossRef CAS PubMed.
- J. R. Heath, C. M. Knobler and D. V. Leff, J. Phys. Chem. B, 1997, 101, 189–197 CrossRef CAS.
- E. Rabani, D. R. Reichman, P. L. Geissler and L. E. Brus, Nature, 2003, 426, 271–274 CrossRef CAS PubMed.
- T. P. Bigioni, X. M. Lin, T. T. Nguyen, E. I. Corwin, T. A. Witten and H. M. Jaeger, Nat. Mater., 2006, 5, 265–270 CrossRef CAS PubMed.
- K. E. Mueggenburg, X. M. Lin, R. H. Goldsmith and H. M. Jaeger, Nat. Mater., 2007, 6, 656–660 CrossRef CAS PubMed.
- R. J. Hickey, Q. J. Luo and S. J. Park, Acs Macro. Lett., 2013, 2, 805–808 CrossRef CAS.
- Y. Y. Geng, M. Liu, K. Tong, J. Xu, Y. I. Lee, J. C. Hao and H. G. Liu, Langmuir, 2014, 30, 2178–2187 CrossRef CAS PubMed.
- Y. A. Liu, L. F. Chen, Y. Y. Geng, Y. I. Lee, Y. Li, J. C. Hao and H. G. Liu, J. Colloid Interface Sci., 2013, 407, 225–235 CrossRef CAS PubMed.
- P. Pieranski, Phys. Rev. Lett., 1980, 45, 569–570 CrossRef CAS.
- Y. Lin, H. Skaff, T. Emrick, A. D. Dinsmore and T. P. Russell, Science, 2003, 299, 226–229 CrossRef CAS PubMed.
- H. W. Duan, D. Y. Wang, D. G. Kurth and H. Mohwald, Angew. Chem., Int. Ed., 2004, 43, 5639–5642 CrossRef CAS PubMed.
- J. B. Pang, S. S. Xiong, F. Jaeckel, Z. C. Sun, D. Dunphy and C. J. Brinker, J. Am. Chem. Soc., 2008, 130, 3284–3285 CrossRef CAS PubMed.
- Y. Lin, H. Skaff, A. Boker, A. D. Dinsmore, T. Emrick and T. P. Russell, J. Am. Chem. Soc., 2003, 125, 12690–12691 CrossRef CAS PubMed.
- S. Gupta, Q. L. Zhang, T. Emrick, A. C. Balazs and T. P. Russell, Nat. Mater., 2006, 5, 229–233 CrossRef CAS.
- W. A. Lopes and H. M. Jaeger, Nature, 2001, 414, 735–738 CrossRef CAS PubMed.
- P. Reiss, E. Couderc, J. De Girolamo and A. Pron, Nanoscale, 2011, 3, 446–489 RSC.
- N. Hundt, H. Quan, N. Hien, P. Sista, J. Hao, J. Servello, K. Palaniappan, M. Alemseghed, M. C. Biewer and M. C. Stefan, Macromol. Rapid Commun., 2011, 32, 302–308 CrossRef CAS PubMed.
- K. K. Huang, Y. K. Fang, J. C. Hsu, C. C. Kuo, W. H. Chang and W. C. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 147–155 CrossRef CAS PubMed.
- M. G. Alemseghed, J. Servello, N. Hundt, P. Sista, M. C. Biewer and M. C. Stefan, Macromol. Chem. Phys., 2010, 211, 1291–1297 CrossRef CAS PubMed.
- M. G. Alemseghed, S. Gowrisanker, J. Servello and M. C. Stefan, Macromol. Chem. Phys., 2009, 210, 2007–2014 CrossRef CAS PubMed.
- E. Schwartz, M. Koepf, H. J. Kitto, R. J. M. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33–47 RSC.
- Z. Q. Wu, K. Nagai, M. Banno, K. Okoshi, K. Onitsuka and E. Yashima, J. Am. Chem. Soc., 2009, 131, 6708–6718 CrossRef CAS PubMed.
- Y. X. Xue, Y. Y. Zhu, L. M. Gao, X. Y. He, N. Liu, W. Y. Zhang, J. Yin, Y. Ding, H. Zhou and Z. Q. Wu, J. Am. Chem. Soc., 2014, 136, 4706–4713 CrossRef CAS PubMed.
- N. Liu, C. G. Qi, Y. Wang, D. F. Liu, J. Yin, Y. Y. Zhu and Z. Q. Wu, Macromolecules, 2013, 46, 7753–7758 CrossRef CAS.
- Y. Y. Zhu, T. T. Yin, J. Yin, N. Liu, Z. P. Yu, Y. W. Zhu, Y. S. Ding, J. Yin and Z. Q. Wu, RSC Adv., 2014, 4, 40241–40250 RSC.
- Z. Q. Wu, C. G. Qi, N. Liu, Y. Wang, J. Yin, Y. Y. Zhu, L. Z. Qiu and H. B. Lu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2939–2947 CrossRef CAS PubMed.
- H. Duan, D. Chen, M. Jiang, W. Gan, S. Li, M. Wang and J. Gong, J. Am. Chem. Soc., 2001, 123, 12097–12098 CrossRef CAS.
- M. Wang, G. Z. Zhang, D. Y. Chen, M. Jiang and S. Y. Liu, Macromolecules, 2001, 34, 7172–7178 CrossRef CAS.
- S. Liu, G. Zhang and M. Jiang, Polymer, 1999, 40, 5449–5453 CrossRef CAS.
- M. Jiang, M. Li, M. L. Xiang and H. Zhou, Adv. Polym. Sci., 1999, 146, 121–196 CrossRef CAS.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew. Chem., Int. Ed., 2001, 40, 3240–3242 CrossRef.
- L. Wang, Y. Fu, Z. Wang, Y. Fan and X. Zhang, Langmuir, 1999, 15, 1360–1363 CrossRef CAS.
- J. R. Kalluri, T. Arbneshi, S. A. Khan, A. Neely, P. Candice, B. Varisli, M. Washington, S. McAfee, B. Robinson, S. Banerjee, A. K. Singh, D. Senapati and P. C. Ray, Angew. Chem., Int. Ed., 2009, 48, 9668–9671 CrossRef CAS PubMed.
- W. T. Lu, A. K. Singh, S. A. Khan, D. Senapati, H. T. Yu and P. C. Ray, J. Am. Chem. Soc., 2010, 132, 18103–18114 CrossRef CAS PubMed.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- J. B. Song, J. J. Zhou and H. W. Duan, J. Am. Chem. Soc., 2012, 134, 13458–13469 CrossRef CAS PubMed.
- W. Li, Y.-G. He, S.-Y. Shi, N. Liu, Y.-Y. Zhu, Y.-S. Ding, J. Yin and Z.-Q. Wu, Polym. Chem., 2015, 6, 2348–2355 RSC.
- S. F. Chong, R. Chandrawati, B. Stadler, J. Park, J. Cho, Y. J. Wang, Z. F. Jia, V. Bulmus, T. P. Davis, A. N. Zelikin and F. Caruso, Small, 2009, 5, 2601–2610 CrossRef CAS PubMed.
- J. H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- W. G. Xu, X. Ling, J. Q. Xiao, M. S. Dresselhaus, J. Kong, H. X. Xu, Z. F. Liu and J. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9281–9286 CrossRef CAS PubMed.
- L. B. Zhong, J. Yin, Y. M. Zheng, Q. Liu, X. X. Cheng and F. H. Luo, Anal. Chem., 2014, 86, 6262–6267 CrossRef CAS PubMed.
- X. M. Qian, X. Zhou and S. M. Nie, J. Am. Chem. Soc., 2008, 130, 14934–14935 CrossRef PubMed.
- H. Gehan, L. Fillaud, M. M. Chehimi, J. Aubard, A. Hohenau, N. Felidj and C. Mangeney, Acs Nano, 2010, 4, 6491–6500 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05430c |
| This journal is © The Royal Society of Chemistry 2015 |