
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pair of Schiff base enantiomers studied by absorption, fluorescence, electronic and vibrational circular dichroism spectroscopies and density functional theory calculation†
Hui-Hui
Tang
,
Lu
Zhang
,
Li-Li
Zeng
,
Xue-Ming
Fang
,
Li-Rong
Lin
* and
Hui
Zhang
College of Chemistry and Chemical Engineering, The University of Xiamen, Xiamen, 361005, P. R. China. E-mail: linlr@xmu.edu.cn
First published on 31st March 2015
Abstract
2-(((1R,2S)-2-Hydroxy-1,2-diphenylethylimino)methyl)phenol and 2-(((1S,2R)-2-hydroxy-1,2-diphenylethylimino)methyl)phenol have been synthesized as a pair of enantiomeric Schiff bases of a chiral salicylaldehyde. These compounds were subsequently characterized by melting point, EI-MS, IR, 1H-NMR and X-ray crystallographic analyses. The UV-vis absorption, fluorescent emission, electronic and vibrational circular dichroism (ECD and VCD) spectral properties of these enantiomers were determined in solution in a variety of different solvents, including acetonitrile, ethanol and hexane, as well as being determined in the solid state. The effects of the different solvents were evaluated in detail, and it was found that the enol-imine tautomer existed as the dominant species in nonpolar solvents, such as hexane, and that the enol-imine and keto-enamine tautomers coexisted in polar solvents, such as ethanol. Theoretical IR and VCD spectra of the enantiomers were calculated at the B3LYP/6-311+G(d,p) level by density functional theory. Given that the different tautomers coexisted in solution and VCD is very sensitive to conformational changes, it was not possible to reliably determine the absolute configurations of the enantiomers based on their solution phase VCD spectra. However, the calculated IR and VCD spectra in a vacuum were in good agreement with the experimental solid state spectra, and it was therefore possible to reasonably assign the absolute configurations of the enantiomers based on the VCD calculations. This study therefore represents a good example of the practical application of solid state VCD spectra to assign the absolute configurations of different enantiomers.
Introduction
Schiff bases have attracted considerable interest from researchers working in a variety of different fields, not only because of their interesting chemical and biological properties, such as their catalytic, bactericidal, flotation and anti-HIV applications, but also because of the potential applications of their linear and nonlinear optical properties.1–12 Schiff bases derived from o-hydroxybenzaldehyde can exist in two tautomeric forms, enol-imine (E-form) and keto-enamine (K-form) forms, both in solution and in solid state through intramolecular hydrogen bonding interactions.13–20 The formation of a zwitterionic structure can also occur as a consequence of proton transfer in both the enol-imine and keto-amine tautomers.21,22 It is well known that the tautomeric equilibrium is sensitive to solvents, and organic solvents normally favor the formation of the E-form, whereas aqueous solvents usually favor the formation of both the E- and K-forms. Furthermore, weakly polar solvents usually favor the formation of the E-form over the K-form. Compounds belonging to this structural class also exhibit strong visible fluorescence because of an excited state intramolecular proton transfer when they change from their E-form to their K-form. Although Schiff bases have been prepared and studied for decades, in terms of their application to the field of photochemical research, most of the compounds studied in this area have been nonchiral compounds. Chiral Schiff bases have been studied to a much greater extent in the field of catalysis, but the photochemical properties of chiral Schiff bases remain largely unexplored. Inspired by the photochemistry of chiral Schiff bases, we recently synthesized 2-(((1R,2S)-2-hydroxy-1,2-diphenylethylimino)methyl)phenol and 2-(((1S,2R)-2-hydroxy-1,2-diphenylethylimino)methyl)phenol as a pair of enantiomeric Schiff bases of a chiral salicylaldehyde. Herein, we report the preparation of these compounds and evaluate their intramolecular proton transfer fluorescence and chiral properties, with particular emphasis on their experimental and calculated IR and VCD spectra.Results and discussion
Crystal structure
R,S-1 crystallized as an orthorhombic system with space group P212121. The molecule was found to exist in its phenol-imine tautomeric form in the crystal structure (Fig. 1). The C22![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N1 and N1–C3 bond distances were 1.281(2) and 1.465(2) Å, respectively. The crystal structure also revealed that R,S-1 adopted an E configuration with respect to the imine CN double bond, with a C16–C22–N1–C1 torsion angle of 173.8(1)°. This result therefore indicated that the benzene ring of the salicylaldehyde moiety was almost coplanar with the imine group. The length of the C21–O2 bond in the crystal structure of R,S-1 was found to be 1.364(2) Å, which suggested that this compound existed as the phenol-imine tautomer in the crystal structure. This contraction of the C22N1 bond was also consistent with the presence of the phenol-imine tautomer. The intramolecular (i.e., O2–H⋯N1) and intermolecular (i.e., O1–H⋯O2) hydrogen bond lengths between O2⋯N1 and O1⋯O2 were found to be 2.549(2) and 2.854(2) Å, respectively. C–H⋯π stacking interactions were also observed between the C11–H bond and the phenyl ring composed of C3–C4–C5–C6–C7–C8 carbons, as well as the C20–H bond and the phenyl ring composed of C16–C17–C18–C19–C20–C21 carbons, with vertical distances of 3.486(3) and 3.471(2) Å, respectively. These intermolecular hydrogen bonding interactions and C–H⋯π stacking interactions led to the direct self-assembly of these molecules into a supramolecular 3D structure (Fig. 1b).
N1 and N1–C3 bond distances were 1.281(2) and 1.465(2) Å, respectively. The crystal structure also revealed that R,S-1 adopted an E configuration with respect to the imine CN double bond, with a C16–C22–N1–C1 torsion angle of 173.8(1)°. This result therefore indicated that the benzene ring of the salicylaldehyde moiety was almost coplanar with the imine group. The length of the C21–O2 bond in the crystal structure of R,S-1 was found to be 1.364(2) Å, which suggested that this compound existed as the phenol-imine tautomer in the crystal structure. This contraction of the C22N1 bond was also consistent with the presence of the phenol-imine tautomer. The intramolecular (i.e., O2–H⋯N1) and intermolecular (i.e., O1–H⋯O2) hydrogen bond lengths between O2⋯N1 and O1⋯O2 were found to be 2.549(2) and 2.854(2) Å, respectively. C–H⋯π stacking interactions were also observed between the C11–H bond and the phenyl ring composed of C3–C4–C5–C6–C7–C8 carbons, as well as the C20–H bond and the phenyl ring composed of C16–C17–C18–C19–C20–C21 carbons, with vertical distances of 3.486(3) and 3.471(2) Å, respectively. These intermolecular hydrogen bonding interactions and C–H⋯π stacking interactions led to the direct self-assembly of these molecules into a supramolecular 3D structure (Fig. 1b).
 | ||
| Fig. 1 Crystal structure of R,S-1 showing the atom-labeling scheme (displacement ellipsoids are drawn at the 50% probability level) (a) and the intramolecular hydrogen-bonds and C–H⋯π interactions (dotted lines) (symmetry codes: (i): x, 1 + y, z, (ii): 1/2 + x, 3/2 − y, −z) (b). | ||
UV-vis spectral characteristics
The UV-vis absorption spectra of R,S-1 and S,R-1 were recorded in the presence of several different solvents, and the results are shown in Fig. 2. It is clear from Fig. 2 that the title compounds afforded characteristic absorption bands at wavelengths in the range of 200–450 nm. The molar absorption coefficient of the band around 200–300 nm was high, with values going as high as 104 L mol−1 cm−1, which were indicative of the π–π* state transition characteristics of the system. A weak n–π* absorption band was observed at 402 nm when the spectra of R,S-1 and S,R-1 were recorded in ethanol, although this band was not observed when the spectra were recorded in acetonitrile or hexane. This result indicated that the weak n–π* absorption band observed in ethanol was caused by the presence of the protic solvent. The peak observed at 402 nm in the UV-vis spectra of R,S-1 and S,R-1 recorded in ethanol was therefore attributed to its K-form.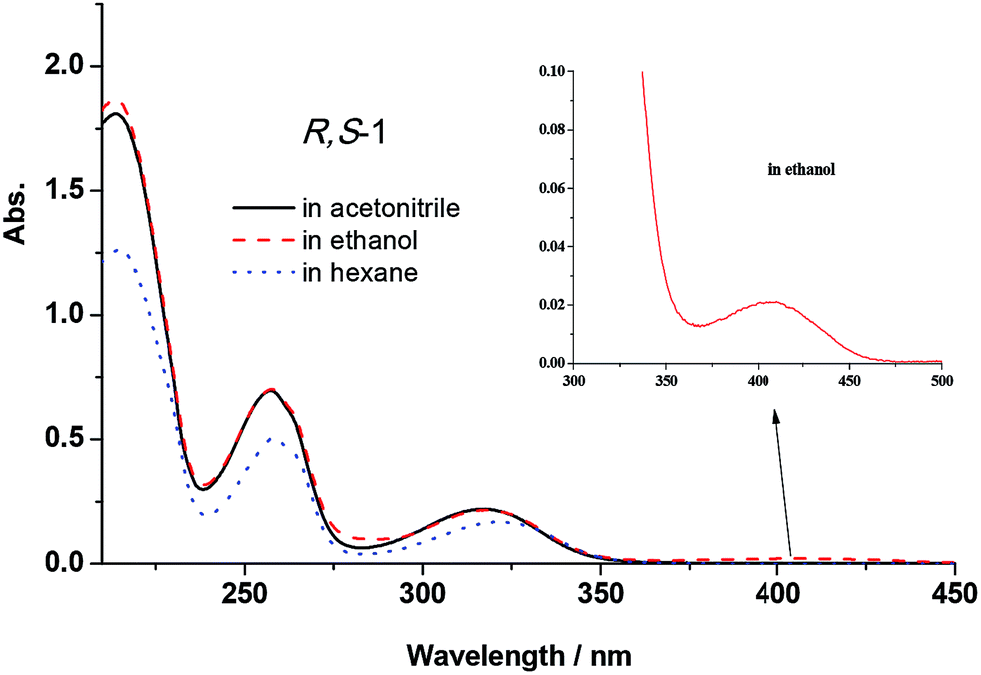 | ||
| Fig. 2 UV-vis absorption spectra of R,S-1 (5 × 10−5 mol L−1) in acetonitrile, ethanol and hexane (the spectra recorded for S,R-1 were the same as shown for R,S-1). | ||
Fluorescence spectral analysis
The fluorescence spectra of R,S-1 and S,R-1 were recorded in a series of different solvents of different polarities, and the results are shown in Fig. 3. The fluorescence intensities of R,S-1 and S,R-1 in ethanol were stronger than those recorded in acetonitrile or hexane at the same concentration. Acetonitrile and ethanol solutions of R,S-1 and S,R-1 emitted locally excited state fluorescence at 360 nm and excited state proton transfer fluorescence at 450 nm. However, the fluorescence spectra of the hexane solutions of R,S-1 and S,R-1 did not show a fluorescence emission at 450 nm.20 The excited state proton transfer fluorescence emissions observed at 450 nm in the fluorescence spectra of R,S-1 and S,R-1 in acetonitrile and ethanol were attributed to emissions from the K-form tautomers of these compounds.23 Hence, the use of a polar protic solvent favored the excited state proton transfer fluorescence emission of R,S-1 and S,R-1. For example, the fluorescence spectra of R,S-1 and S,R-1 in ethanol gave particularly strong excited state proton transfer fluorescence emissions at 450 nm. These results therefore suggested that the use of a protic solvent favored the formation of the K-form tautomers of these compounds, as indicated by the strong fluorescence emissions at 450 nm in their spectra. The ground state geometry of R,S-1 in acetonitrile was optimized using a DFT(B3LYP) method with a 6-311+G(d,p) basis set. The frontier molecular orbitals, which are commonly referred to as the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals, are the key parameter in determining the molecular properties, especially the molecular electrical transport properties.24 The plotting of the HOMO and LUMO of R,S-1 (Fig. 4) revealed that the HOMO consisted of a π orbital, which was mainly localized on the salicylaldehyde imine moiety. Furthermore, the LUMO was found to be predominantly localized on the anti-bonding π orbital of the salicylaldehyde group. This result indicated that the excited state of R,S-1 (or S,R-1) would accommodate the intramolecular charge transfer more readily than the ground state. Hence, although the weak n–π* absorption band was difficult to observe in acetonitrile, the excited state proton transfer fluorescence emissions can be still observed at 450 nm in the fluorescence spectra of R,S-1 and S,R-1 in acetonitrile solution. This observation is due to the presence of a small amount of keto tautomers in the polar, non-protic acetonitrile solution. | ||
| Fig. 3 Fluorescence spectra of R,S-1 (5 × 10−5 mol L−1) in acetonitrile, ethanol and hexane (the spectra recorded for S,R-1 were the same as shown for R,S-1). | ||
 | ||
| Fig. 4 Frontier molecular orbitals of R,S-1 at the B3LYP/6-311+G(d,p) level. | ||
Electronic circular dichroism spectral properties
Fig. 5 shows the electronic dichroism (ECD) spectra of R,S-1 and S,R-1 in acetonitrile, ethanol and hexane solutions, as well as their spectra in the solid state as KBr pellets. The ECD spectra of the two enantiomers were perfect mirror-images of each other, both in solution and the solid state. Compared with their UV-vis spectra, the ECD spectra of R,S-1 and S,R-1 revealed typical negative and positive excitation splitting patterns around 257 nm. Furthermore, the peak absorption in the ECD spectra at 318 nm was very weak in solution and much stronger in the solid state. This peak in the absorption at 318 nm was attributed the enol forms of the two compounds, as evidenced by their UV-vis spectra. The strong absorption at 318 nm in the ECD spectra of the solid state samples could be attributed to the hindered rotation of the CN double bond attached to the phenyl ring carbon in the KBr pellet. A separate absorption band was also observed in the ECD spectra at 415 nm, which was attributed to the n–π* absorption transition of the CN bond in the solid state.
 | ||
Fig. 5 ECD spectra of R,S-1 and S,R-1 (5 × 10−5 mol L−1) as solutions in acetonitrile, ethanol and hexane, as well as in the solid state as KBr pellets (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200). :200). | ||
Vibrational circular dichroism spectral properties
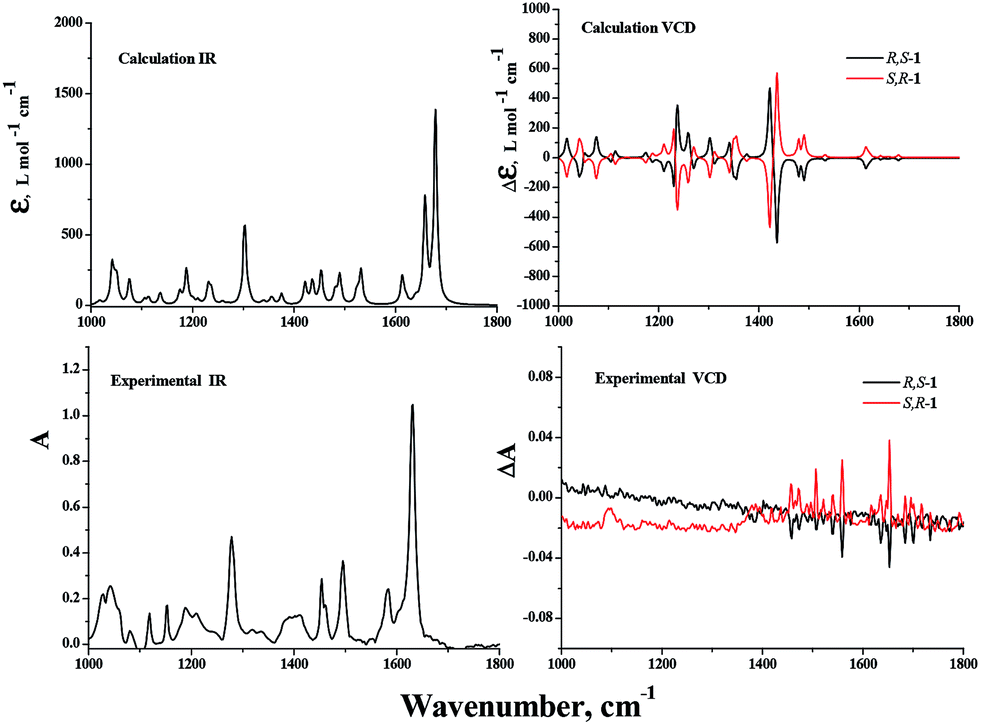
Vibrational circular dichroism (VCD) spectroscopy has been shown to be a leading tool for the investigation of conformational preferences and solvent effects for a variety of materials both in solution and in the solid state and today it is frequently applied to the determination of absolute configurations.25–29 In our effort, the VCD and IR absorption spectra of R,S-1 and S,R-1 were measured in solution in CDCl3, as well as in the solid state as KBr pellets. The VCD spectra of R,S-1 and S,R-1 in CDCl3 contained a few very weak bands, as shown in Fig. 6. It is clear from the results in Fig. 6 that there were slight signals between 1450–1650 cm−1 and the two enantiomers gave equal and opposite signals for this band. If the bands observed in the region 1450–1650 cm−1 can be assigned to robust vibrational modes, their signs are less likely to be changed by small perturbations present in the experiment or by using different computational parameters, it would be possible to reliably determine the absolute configuration of this system based on these weak VCD signals.30,31 However, it is not possible to reliably determine the absolute configuration of a system based on weak VCD signals, if these signs are originated from the rotational strength of non-robust modes.31,32 Although the VCD sign of robust modes can be accurately predicted from calculation and can be trusted to use to establish the absolute configuration, VCD spectra recorded in solution are generally compared with the Boltzmann population weighted composite of the calculated spectra of some of the lowest possible conformers.33 The computational workload required for this process can be particularly heavy, especially when the molecule under consideration can exist in a variety of different conformations. With this in mind, we tried to measure the solid phase VCD spectra of the two enantiomers and compare these results with those of the calculated VCD spectra in a vacuum. Pleasingly, the experimental and theoretical calculations were found to be in good agreement. The VCD spectra of R,S-1 and S,R-1 measured as KBr pellets are shown in Fig. 7. Apparently, the selected wavelength region provides rich spectra with well resolved vibrational transitions. The R,S-1 and S,R-1 enantiomers gave identical IR spectra, and their VCD spectra were almost symmetrical, mirror-images of each other with respect to the baseline. The results in Fig. 7 clearly show that there was a reasonable degree of similarity between the observed and simulated IR and VCD patterns. The absorption peak calculated at 1647 cm−1 and subsequently observed at 1627 cm−1 corresponds to the stretching vibration of the CN bond. The sign of this band can be either positive or negative depending on the nature of the enantiomer. Its angle between the electric and magnetic vibrational transitional dipolar moment is close to 0 or 180 degrees. The VCD sign of this band can be characterized as a robust vibrational mode and therefore should be trusted.30 The peak seen at 1580 cm−1 in the experimental IR spectrum was attributed to an aromatic CC stretching vibration. However, this peak was weak in the calculated IR and VCD spectra, as well as the experimental VCD spectrum. The calculated VCD peak from 1480 to 1350 cm−1 can correspond to the bands observed between 1510 and 1400 cm−1 for the vibrational mode of the phenyl ring. It is noteworthy that the VCD bands in the range of 1100 to 1350 cm−1 were attributed to the C–O stretching and out-of-plane OH bending vibrations. The calculated spectrum did not contain a positive–negative couplet around 1150 cm−1 for the C–O stretching vibration, but did contain VCD couplets in the range of 1200 to 1350 cm−1. The band around 1150 cm−1 was assigned to the C–O stretching vibration of the phenol furthest away from the chiral center, whereas the VCD couplets in the range of 1200 to 1350 cm−1 were assigned to the C–O stretching vibration of the chiral hydroxyl center. The calculated bands around 915 cm−1 for the out-of-plane CH bending vibrations of the phenyl ring were found to be weak in the experimental VCD spectra, most likely because they were far from the chiral center. A comparison of the calculated and observed VCD spectra revealed that the absorption peaks involving the chiral center and group vibrations could be assumed relative to the position that the chiral center appeared in the experimental VCD spectra. The theoretical calculation of the IR and VCD spectra could therefore provide results in good agreement with the experimental observations, which would allow for the absolute configurations of the synthesized enantiomers to be reasonably assigned according to the theoretical VCD calculation. This is therefore a good example of the practical application of solid state VCD spectra to assist in assigning the absolute configurations of a pair of synthesized enantiomers according to their experimental and calculated VCD spectra.
 | ||
| Fig. 6 DFT calculated IR and VCD spectra, as well as the experimental IR and VCD spectra, of R,S-1 and S,R-1 in CDCl3. | ||
 | ||
| Fig. 7 DFT calculated IR and VCD spectra of R,S-1 and S,R-1 in the gas phase, and the experimental IR and VCD spectra of R,S-1 and S,R-1 in the solid state as KBr pellets (1:50). | ||
Experimental section
Reagents
Enantiopure (1R,2S)-(−)-2-amino-1,2-diphenylethanol and (1S,2R)-(+)-2-amino-1,2-diphenylethanol were purchased from Shanghai Darui Fine Chemicals Ltd. Co. The other chemicals were obtained from Sinopharm Chemical Reagent Ltd. Co. and used as received without further purification. Analytical-grade solvents were all received from Shanghai Chemicals Group Co. and were redistilled before use. Distilled–deionised water was used throughout the experiment.Apparatus
Absorption spectra were obtained using a Shimadzu UV224012PC absorption spectrophotometer. Fluorescence spectra were obtained using a Hitachi F-7000 spectrofluorometer. The solid-state infrared spectra were recorded on a Nicolet AVATAR FT-IR330 spectrometer by using pressed KBr pellets. 1H NMR spectra were acquired on a Bruker Unity 400 MHz spectrometer using TMS as an internal standard. EI-MS data were obtained on a Bruker ESQUIRE-3000 Plus LC-MS/MS spectrometer. Electronic circular dichroism (ECD) spectra were recorded with a JASCO J-810 spectropolarimeter. Vibrational circular dichroism (VCD) spectra were scanned on a BioTools ChiralIR-2X spectrometer. The melting point was determined with an X-4 micromelting point apparatus without correction. The X-ray diffraction data were collected on a Bruker SMART Apex CCD diffractometer equipped with graphite-monochromatic Mo Kα (λ = 0.71073 Å) radiation using an ω scan mode at 293(2) K.Physical measurements
A cell was filled with 3.0 mL of the Schiff base and a variety of different solvents at the same concentration, and the UV, emission and ECD spectra of the resulting solutions were then collected. The size of the slit was set at 1 nm for the UV and electronic dichroism (ECD) spectra measurements. The fluorescence emission intensity was measured at an excitation wavelength of 300 nm. Spectral bandwidths for the excitation and emission monochromators were set at 5 nm. The solid state transmission ECD spectra were recorded at wavelengths in the range of 500–200 nm at room temperature using the Pellet CD Attachment. All of the spectra were recorded at a scanning speed of 100 nm min−1, with a step size of 0.2 nm, a bandwidth of 2 nm, a response time of 0.5 s and an accumulation of five scans. All of the spectra were background corrected. Solid-phase samples were prepared using the KBr pellet technique (1.0 mg of the title compound per 200 mg of KBr). Crystalline samples were mixed with dried KBr, finely ground in a SpecaMill for 10 min and then pressed with 15 ton of pressure under a vacuum for 3 min to make a spotless, transparent disk with a diameter of 12 mm. The individual pellets were mounted on a rotatable holder in the spectropolarimeter. Several ECD spectra were measured for each sample by rotating the disk around the incident axis at a variety of different rotation angles to guarantee identical spectra and the absence of detectable spectral artifacts.34,35 VCD spectra were recorded at a resolution of 4 cm−1 using Synchrocell (2.75 s per cycle). The ZnSe photoelastic modulator of the instrument was set to 1400 cm−1. To improve the S/N ratio, the spectra were measured for 15 h. Solution VCD spectra were scanned using a CDCl3 solution of the compound (ca. 0.02–0.05 M), which was injected into a cell (150 μm in optical length) with BaF2 windows. The correction performed involved the subtraction of the solvent spectra. Solid state samples were prepared using the KBr pellet technique (using 1.0 mg of the compound per 50 mg of KBr, this higher concentration did not cause the appearance of artifacts in the recorded IR spectra). A baseline correction was achieved by subtracting the spectrum of a reference KBr pellet that had been collected under the same conditions. Additionally, fifteen VCD spectra for each sample were collected over 15 h of measurements. The final spectra were obtained by averaging the 15 spectra and subtracting the spectrum of a reference KBr pellet that had been collected under the same conditions. The final baseline spectrum was also obtained by averaging the 15 spectra over 15 h of measurement. The crystal data integration and reduction steps were performed using the SAINT software. An empirical absorption correction was applied to the collected reflections using SADABS. The crystal structure was solved using direct methods with SHELXS-97 and refined by full-matrix least-squares calculations with SHELXL-97 based on F2.36 All non-hydrogen atoms were located at their calculated positions. Graphics were generated using Diamond 3.0 for Windows.Theory calculations
Density functional theory (DFT) closed shell calculations were performed with the B3LYP hybrid functional with a 6-311+G(d,p) basis set using the Gaussian 09 suite of programs37 at the College of Chemistry and Chemical Engineering. The conformer resulting from the crystal structure was used for optimization. Complete optimization was performed using all of the internal coordinates of the molecule. VCD computations were performed for individual molecules both in a vacuum and in CHCl3 solvent. The solvent was described using the polarizable continuum model. Calculated frequencies were empirically scaled using a 0.96 scaling factor. MO plots were drawn using the GaussView software.Preparation of 2-(((1R,2S)-2-hydroxy-1,2-diphenylethylimino)methyl)phenol (R,S-1) and 2-(((1S,2R)-2-hydroxy-1,2-diphenylethylimino)methyl)phenol (S,R-1)
(1R,2S)-(−)-2-Amino-1,2-diphenylethanol (1.06 g, 5 mmol) and salicylaldehyde (1.0 mL, 10 mmol) were dissolved in methanol (50 mL), and the resulting solution was heated at 60 °C for 8 h. The solution was then cooled to room temperature to give a yellow precipitate, which was collected by filtration under reduced pressure. The solid material was collected and purified by column chromatography eluting with a 3:1 (v/v) mixture of petroleum and ethyl acetate to afford R,S-1 as a yellow powder (1.12 g, 78%). The pure product was dissolved in a 1:1 (v/v) mixture of ethanol and acetonitrile, and the resulting solution was filtered into a test tube before being allowed to slowly evaporate at room temperature. This process afforded yellow crystals suitable for X-ray crystallography analysis within 7 days. M.P. 125–126 °C; EI-MS: found 318.2, calcd for C21O2NH19 + H+: 318.3; IR (ν, cm−1): 3447, 3058, 3029, 2873, 1629, 1578, 1499, 1453, 1425, 1343, 1276, 1217, 1201, 1115, 1084, 1054, 1026, 986, 900, 854, 785, 771, 709, 696; 1H NMR (DMSO-d6, 400 MHz) δ [ppm]: 13.33 (s, 1H, OH), 8.37 (d, J = 4.4 Hz, 1H, CH), 7.35–7.11 (m, 12H, ArH), 6.84 (dd, J = 11.2, 4.4 Hz, 2H), 5.63 (d, J = 4.5 Hz, 1H, OH), 4.99 (dd, J = 6.2, 4.6 Hz, 1H, CH), 4.64 (d, J = 6.3 Hz, 1H, CH) (the preparation and characterization data of S,R-1 were the same as those of R,S-1).
Conclusion
In summary, we have prepared 2-(((1R,2S)-2-hydroxy-1,2-diphenylethylimino)methyl)phenol (R,S-1) and 2-(((1S,2R)-2-hydroxy-1,2-diphenylethylimino)methyl)phenol (S,R-1) as a pair of enantiomeric Schiff bases of a chiral salicylaldehyde. These compounds were characterized by melting point, EI-MS, IR, 1H-NMR and X-ray crystallographic analysis. The UV-vis absorption, fluorescent emission and circular dichroism spectral properties of these enantiomers were measured in solution in acetonitrile, ethanol and hexane, and the results were subjected to detailed analysis. The results revealed that the enol-imine tautomers of the enantiomers were the dominant species in nonpolar solvents, such as hexane, and that the enol-imine and keto-enamine tautomers coexisted in polar solvents, such as ethanol. The R,S-1 and S,R-1 enantiomers gave identical IR spectra, and their solid state VCD spectra were almost symmetrical, mirror-images of each other. The absorption peaks involving the chiral center and group vibrations could be related to the position of the chiral center in the experimental VCD spectrum. Theoretical calculations of the IR and VCD spectra were found to be in good agreement with the experimental observations for the solid state spectra. The absolute configurations of the synthesized enantiomers could therefore be reasonably assigned based on the VCD calculations. Our structural analyses of the present molecules may be taken as an example to illustrate those chiral molecules which have many isomers in solution as an alternative to using solid state VCD spectra to analyze their absolute configuration.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (grant nos 21271150 and 21273175) and NFFTBS (grant no. J1310024).Notes and references
- S. Mouri, Z. Chen, H. Mitsunuma, M. Furutachi, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 1255–1257 CrossRef CAS PubMed.
- G. G. Mohamed, M. M. Omar and A. M. Hindy, Spectrochim. Acta, Part A, 2005, 62, 1140–1150 CrossRef PubMed.
- R. B. Sumathi and M. B. Halli, Bioinorg. Chem. Appl., 2014, 2014, 942162 CAS.
- G. Cerchiaro, K. Aquilano, G. Filomeni, G. Rotilio, M. R. Ciriolo and A. M. Ferreira, J. Inorg. Biochem., 2005, 99, 1433–1440 CrossRef CAS PubMed.
- N. S. Geweely, Arch. Microbiol., 2009, 191, 687–695 CrossRef CAS PubMed.
- S. Gopalakrishnan and J. Joseph, Mycobiology, 2009, 37, 141–146 CrossRef CAS PubMed.
- R. S. Joseyphus and M. S. Nair, Mycobiology, 2008, 36, 93–98 CrossRef CAS PubMed.
- T. Rosu, E. Pahontu, D. C. Ilies, R. Georgescu, M. Mocanu, M. Leabu, S. Shova and A. Gulea, Eur. J. Med. Chem., 2012, 53, 380–389 CrossRef CAS PubMed.
- A. Trujillo, M. Fuentealba, D. Carrillo, C. Manzur, I. Ledoux-Rak, J. R. Hamon and J. Y. Saillard, Inorg. Chem., 2010, 49, 2750–2764 CrossRef CAS PubMed.
- M. Sun, Y. Ding, G. Cui and Y. Liu, J. Phys. Chem. A, 2007, 111, 2946–2950 CrossRef CAS PubMed.
- J. S. Rani and V. Ramakrishnan, Spectrochim. Acta, Part A, 2013, 114, 170–174 CrossRef PubMed.
- S. S. Al-Jaroudi, M. Monim-Ul-Mehboob, M. Altaf, A. A. Al-Saadi, M. I. Wazeer, S. Altuwaijri and A. A. Isab, BioMetals, 2014, 27, 1115–1136 CrossRef CAS PubMed.
- J. M. Ortiz-Sanchez, R. Gelabert, M. Moreno and J. M. Lluch, J. Phys. Chem. A, 2006, 110, 4649–4656 CrossRef CAS PubMed.
- M. Rubcic, K. Uzarevic, I. Halasz, N. Bregovic, M. Malis, I. Dilovic, Z. Kokan, R. S. Stein, R. E. Dinnebier and V. Tomisic, Chemistry, 2012, 18, 5620–5631 CrossRef CAS PubMed.
- S. Sharif, G. S. Denisov, M. D. Toney and H. H. Limbach, J. Am. Chem. Soc., 2006, 128, 3375–3387 CrossRef CAS PubMed.
- D. Guha, A. Mandal, A. Koll, A. Filarowski and S. Mukherjee, Spectrochim. Acta, Part A, 2000, 56, 2669–2677 CrossRef CAS.
- A. Mandal, D. Fitzmaurice, E. Waghorne, A. Koll, A. Filarowski, S. Quinn and S. Mukherjee, Spectrochim. Acta, Part A, 2004, 60, 805–813 CrossRef CAS.
- J. M. Ortiz-Sanchez, R. Gelabert, M. Moreno and J. M. Lluch, J. Chem. Phys., 2008, 129, 214308 CrossRef PubMed.
- C. Randino, M. Ziolek, R. Gelabert, J. A. Organero, M. Gil, M. Moreno, J. M. Lluch and A. Douhal, Phys. Chem. Chem. Phys., 2011, 13, 14960–14972 RSC.
- M. Ziolek, J. Kubicki, A. Maciejewski, R. Naskrecki and A. Grabowska, J. Chem. Phys., 2006, 124, 124518 CrossRef PubMed.
- M. H. Habibi, E. Shojaee, M. Ranjbar, H. R. Memarian, A. Kanayama and T. Suzuki, Spectrochim. Acta, Part A, 2013, 105, 563–568 CrossRef CAS PubMed.
- H. Karabiyik, B. Guzel, M. Aygun, G. Boga and O. Buyukgungor, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o215–218 CAS.
- W. Rodriguez-Cordoba, J. S. Zugazagoitia, E. Collado-Fregoso and J. Peon, J. Phys. Chem. A, 2007, 111, 6241–6247 CrossRef CAS PubMed.
- K. Fukui, Science, 1982, 218, 747–754 CAS.
- P. J. Stephens, J. Phys. Chem., 1985, 89, 748–752 CrossRef CAS.
- L. A. Nafie, T. A. Keiderling and P. J. Stephens, J. Am. Chem. Soc., 1976, 98, 2715–2723 CrossRef CAS.
- J. Sadlej, J. C. Dobrowolski and J. E. Rode, Chem. Soc. Rev., 2010, 39, 1478–1488 RSC.
- H. Sato, T. Nakae, K. Morimoto and K. Tamura, Org. Biomol. Chem., 2012, 10, 1581–1586 CAS.
- T. Sasaki, I. Hisaki, T. Miyano, N. Tohnai, K. Morimoto, H. Sato, S. Tsuzuki and M. Miyata, Nat. Commun., 2013, 4, 1787–1794 CrossRef PubMed.
- V. P. Nicu and E. J. Baerends, Phys. Chem. Chem. Phys., 2009, 11, 6107–6118 RSC.
- V. P. Nicu, E. Debie, W. Herrebout, B. Van der Veken, P. Bultinck and E. J. Baerends, Chirality, 2009, 21, E287–E297 CrossRef CAS PubMed.
- C. Merten, R. McDonald and Y. J. Xu, Inorg. Chem., 2014, 53, 3177–3182 CrossRef CAS PubMed.
- L. A. Nafie, Vibrational optical activity : principles and applications, Wiley, Chichester, West Sussex, 2011 Search PubMed.
- L. Ding, L. R. Lin, C. Y. Liu, H. K. Li, A. J. Qin, Y. Liu, L. Song, H. Zhang, B. Z. Tang and Y. F. Zhao, New J. Chem., 2011, 35, 1781–1786 RSC.
- R. Kuroda, T. Harada and Y. Shindo, Rev. Sci. Instrum., 2001, 72, 3802–3810 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXS 97 and SHELXL 97, University of Göttingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. B. Scalmani, V. B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † CCDC 992258. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra02154e |
| This journal is © The Royal Society of Chemistry 2015 |