
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled surface mineralization of metal oxides on nanofibers†
Nesrin
Horzum
 ab,
Margherita
Mari
a,
Manfred
Wagner
a,
Giuseppino
Fortunato
c,
Ana-Maria
Popa
c,
Mustafa M.
Demir
d,
Katharina
Landfester
a,
Daniel
Crespy
a and
Rafael
Muñoz-Espí
*a
ab,
Margherita
Mari
a,
Manfred
Wagner
a,
Giuseppino
Fortunato
c,
Ana-Maria
Popa
c,
Mustafa M.
Demir
d,
Katharina
Landfester
a,
Daniel
Crespy
a and
Rafael
Muñoz-Espí
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: munoz@mpip-mainz.mpg.de; Fax: +49 6131 379 100; Tel: +49 6131 379 410
bDepartment of Engineering Sciences, İzmir Katip Çelebi University, Çiğli, 35620 İzmir, Turkey
cEmpa, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland
dDepartment of Material Science and Engineering, İzmir Institute of Technology, Urla, 35430, İzmir, Turkey
First published on 16th April 2015
Abstract
We report a versatile approach for the preparation of metal oxide/polymer hybrid nanofibers by in situ formation of metal oxide nanoparticles on surface-functionalized polymer fibers. Poly (styrene-co-vinylphosphonic acid) fibers were produced by electrospinning and used as supports for the in situ formation of ceria nanocrystals without further thermal treatment. The crystallization of ceria was induced by the addition of an alkaline solution to fibers loaded with the corresponding precursor. The formation of the inorganic material at the fiber surface was investigated by transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS). The extension of the approach to prepare polymer/titania hybrid nanofibers demonstrates its versatility.
1. Introduction
In recent years, materials based on transition metal oxides have played an important role in the development of novel semiconductor materials for a wide range of applications. In this work, we have selected two representative metal oxides: cerium(IV) oxide (ceria) and titanium(IV) oxide (titania). Ceria is a rare-earth oxide material employed as ultraviolet absorbent, gas sensor, photocatalyst, and electrolyte for fuel cells.1 Titania finds applications in photocatalysis, solar energy conversion, sensors, and mesoporous membranes.2A large surface area of metal oxide materials, which is needed for many applications, can be achieved by reducing their size to the nanometric range. However, the high surface energy of nanoparticles causes aggregation during synthesis or post-synthesis processes. To overcome this major issue, support materials are often used to hinder the aggregation of the nanoparticles. Polymer particles have been applied as supports, coating materials, and structure-directing agents to provide heterogeneous nucleation and to control the growth of inorganic materials. Several works have reported the crystallization of metal oxides on the surface of polystyrene particles with grafted anionic or cationic polyelectrolyte brushes. TiO2 nanoparticles were immobilized on polystyrene cores with long chains of poly(styrene sodium sulfonate).3,4 Mixed valent manganese oxide (MnOx) nanoparticles were stabilized by 2-(trimethylammonium)ethyl methacrylate chloride.5 Poly(styrene-co-acetoxyethyl methacrylate) latex particles were also utilized for the in situ crystallization of ZnO,6 Ta2O5,7 TiO2,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and In(OH)39 in alcoholic solvents. Recently, Fischer et al.10 synthesized specifically designed surface-active monomers (so-called surfmers) and applied the surfmer-functionalized polymer particles as nucleation surfaces for the controlled surface precipitation of CeO2, α-Fe2O3, Fe3O4, and ZnO nanoparticles from both alcoholic and aqueous media.
8 and In(OH)39 in alcoholic solvents. Recently, Fischer et al.10 synthesized specifically designed surface-active monomers (so-called surfmers) and applied the surfmer-functionalized polymer particles as nucleation surfaces for the controlled surface precipitation of CeO2, α-Fe2O3, Fe3O4, and ZnO nanoparticles from both alcoholic and aqueous media.
Polymer fibers can also be used as supports to place inorganic particles in a structural template in an isolated and non-aggregated fashion.11,12 The advantage of fiber networks over nanoparticles is that they can be easily separated from the reaction media. The free-standing non-woven possesses large surface area and high porosity that enables surface modification or incorporation of particles in the fibers. In an analogous way to the achievements with polymer particles, tailor-made surface chemistry on the fibers can provide nucleation centers for controlled crystallization. The methods for fabricating functional metal oxide fibers can be classified in three groups: (i) fabrication of electrospun metal oxide fibers by high temperature heat treatment (calcination);13–16 (ii) electrospinning polymer solution containing metal oxide nanoparticles;17–20 and (iii) metal oxide coating of polymer fibers.21–24 Although calcination of polymer/metal oxide precursor fibers is the most common method, fibrous membranes after this process are typically brittle and lose their mechanical integrity.25 In the second case, metal oxide nanoparticles are embedded in polymer fibers that reduce the performance of the available active sites due to reduced surface coverage.26,27 Thus, the fabrication of organic/inorganic hybrids by the in situ formation of metal oxide nanoparticles on the surface of electrospun fibers can be suitable for fabricating hybrid materials, serving also at the same time as an interesting model to understand mineralization processes. This in situ crystallization method provides on one hand the possibility of synthesis at lower temperature without any additional heat treatment, and on the other hand a controlled crystallization on the surface of the non-woven. The successful in situ synthesis of metal oxide nanoparticles on the surface of polymeric fibers, while maintaining the properties of the polymer template, remains very challenging, and only very few related works have been reported.21–24,28,29
Surface treatment of electrospun fibers can prevent particle aggregation and ensure homogeneous distribution of particles on the fibers. Drew et al.21 reported the fabrication of electrospun polyacrylonitrile (PAN) nanofibers coated with metal oxides (TiO2 and SnO2) by liquid phase deposition. The formation of the metal oxide was driven by the immersion of the PAN fibrous membrane in an aqueous solution of the corresponding metal halide salts and halogen scavengers. The same group also reported that PAN nanofibers treated with hot sodium hydroxide could be functionalized with carboxylic groups, which induced fast nucleation of metal oxide growth on the fiber surface.28 Following a different strategy, Nagamine and coworkers22 fabricated hollow TiO2 fibers by electrospinning an aqueous poly(ethylene oxide) solution into a tetraisopropoxide–hexane solution. Employing the same procedure, PVA–TiO2 composite nanofibers were fabricated by modifying the surface nanostructure by alkaline and subsequent acid treatment.29 Zhang et al.24 presented a method for the preparation of TiO2 nanoparticles in electrospun poly(methyl methacrylate) (PMMA). The method involved the electrospinning of a TiO2 precursor–PMMA solution followed by a mild hydrothermal treatment to retain the properties of the polymeric template. Shu and Li23 prepared electrospun cellulose acetate fibers, and the synthesis of TiO2 nanoparticles were achieved on the cellulose fibers by the interfacial interaction between the hydroxyl groups and hydrated Ti(OBu)4.
Here, we present a versatile approach to crystallize in situ inorganic metal oxides, exemplified with the cases of ceria and titania, on the surface of electrospun poly(styrene-co-vinylphosphonic acid) fibers. Thereby, it is suggested that the polymer offers stable and absorption sites for metal oxides at high concentrations.
2. Experimental section
2.1 Materials
Styrene (Merck, 99%) was purified by passing through a column packed with aluminum oxide (Fluka, chromatography). Vinylphosphonic acid (VPA, Aldrich, 97%) was washed with diethyl ether (Aldrich, 99.5%) to remove the inhibitor and dried under vacuum. Sodium dodecyl sulfate (SDS, Carl Roth, 99%), 2,2′-azobis-(2-methylbutyronitrile) (V59, Wako Chemicals, 98%), hexadecane (Aldrich, 99%), cerium(III) nitrate hexahydrate (Fluka, ≥99.0%), titanium isopropoxide (TIP, Aldrich, 99.9%), sodium hydroxide (Riedel de-Haën, 99%), isopropanol (Aldrich, 99.8%), and N,N-dimethylformamide (DMF, Aldrich, 99.8%) were all used as received without any further purification. Deionized water (18.2 MΩ cm) was used throughout the experiments.2.2 Synthesis of phosphonate-functionalized polystyrene by miniemulsion
Polystyrene (PS) and poly(styrene-co-vinylphosphonic acid) (P(S-co-VPA)) aqueous dispersions were synthesized by miniemulsion polymerization. A 0.3 wt% aqueous solution of the surfactant was prepared by dissolving SDS (72 mg) in water (24 g) and used as a continuous phase. The surfactant solution was added to a solution containing styrene (6 g), hexadecane (250 mg), and the initiator V59 (100 mg). Hexadecane was used as a costabilizer to suppress the Ostwald ripening and stabilize the miniemulsions against molecular diffusion. The use of the oil-phase initiator V59 minimizes the formation of water soluble oligomers in the continuous phase.30 The amount of VPA was varied (0, 1, 5, and 10 wt% based on the total monomer amount) for the synthesis of P(S-co-VPA). The resulting polymers are named P0, P0.01, P0.04, and P0.10 respectively, with respect to the molar ratio of VPA. After pre-emulsification of the initial mixture for 1 h at 1000 rpm, the miniemulsion was prepared by ultrasonication for 2 min at 90% amplitude (Branson Sonifier W-450D, 1/2 inch tip) under cooling with an ice-water bath. The miniemulsion was flushed with argon for 10 min to remove oxygen. The polymerization was carried out at 72 °C for 12 h and the polymers were isolated by freeze-drying.2.3 Fabrication of the P(S-co-VPA)/metal oxide hybrid nanofibers
A sample of PS or P(S-co-VPA) was dissolved in DMF by stirring until complete dissolution was achieved. The solid content of the solution was 8 wt%. The viscous solution was loaded in a plastic syringe connected with silicone rubber tubing. The electrospinning experiments were carried out with a commercial platform (ES1a, Electrospinz). The positive electrode was applied to the spinneret and either an aluminum foil or a metal paper clip was used as a grounded counter electrode. Metal paper clips were attached on the foil to prevent folding of the non-woven for the subsequent crystallization process. A potential of 12 kV was applied and the flow rate of the polymer solution was kept at 1.5 mL h−1 by using a syringe pump (Bioblock, Kd Scientific). The tip-to-collector distance was 14 cm and the electrospinning time was fixed to 3 min. The as-prepared electrospun fibers (F0, F0.01, F0.04, and F0.10) were dried for 12 h at 50 °C under vacuum to remove residual solvent.For the crystallization of the ceria nanoparticles on the polymer fibers, the nanofiber coated substrate was immersed into 10 mL of a 0.5 mM Ce(NO3)3·6H2O aqueous solution for 3 h under constant stirring (300 rpm) at room temperature. The Ce3+-loaded polymer non-woven was then separated from the solution. Then, 10 mL of a 1.0 mM NaOH solution was added dropwise with a rate of 1 mL h−1 under constant stirring at 300 rpm. The resulting hybrid fiber was washed with deionized water at least three times and dried under vacuum at 40 °C for 14 h.
To conduct the formation of titania, the electrospun nanofiber-coated substrate was immersed into an isopropanol–H2O mixture (200:1, v/v) under constant shaking (300 rpm) at 40 °C. An isopropanol–titanium isopropoxide mixture (100:1, v/v) was prepared under argon atmosphere and 5 mL of the mixture was added dropwise with a rate of 5 mL h−1 to the solution containing non-woven. The resulting hybrid fiber was isolated from the solution and dried under vacuum at 40 °C for 14 h.
The fibers between the clips were used for morphological observations by scanning electron microscopy (SEM) in a Hitachi SU8000 microscope. The diameter of fibers was statistically estimated from SEM micrographs measuring over more than 50 fibers by using the software Fiji/ImageJ. Thermogravimetric analysis (TGA) was carried out with a Mettler Toledo 851 thermobalance under nitrogen atmosphere and heating rate of 10 °C min−1.
The cross-sectional transmission electron microscopy (TEM) images of the polymer/metal oxide hybrid nanofibers were captured in a FEI Tecnai F-20 microscope. The nanofibers were embedded into epoxy and cut into ultrathin slices using an ultra-microtome (Sorvall MT 5000).
Surface compositions were determined by X-ray photoelectron spectroscopy (XPS) (PHI LS 5600) with a standard Mg-Kα X-ray source. The energy resolution of the spectrometer was set to 0.8 eV per step at pass energy of 187.85 eV for survey scans. The X-ray beam was operated at a current of 25 mA and an acceleration voltage of 13 kV. Charge effects were corrected using carbon C 1s = 284.5 eV. The determination of the concentrations of surface elements was performed using CasaXP software.
3. Results and discussion
Miniemulsion polymerization is an efficient process to synthesize functionalized polymers either through homopolymerization of a functional monomer or its copolymerization with another comonomer. This technique allows the copolymerization of monomers with different polarities such as the hydrophobic styrene with the hydrophilic vinylphosphonic acid.32 The phosphonic groups are able to interact with metal precursors.The general procedure for the fabrication of P(S-co-VPA) by miniemulsion polymerization and electrospun fibers covered by metal oxide nanoparticles is depicted in Fig. 1.
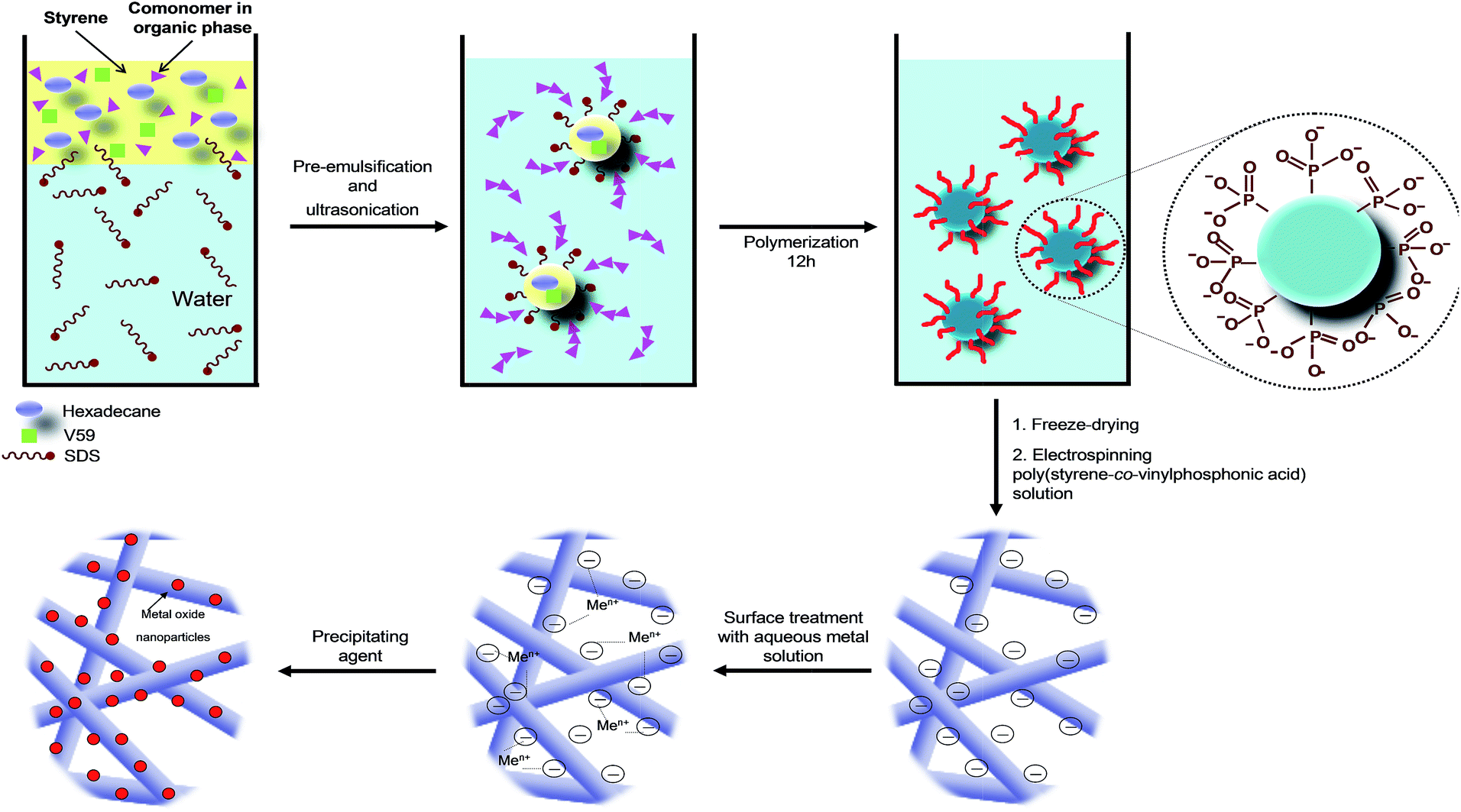 | ||
| Fig. 1 Illustration of miniemulsion polymerization and mechanism of metal oxide formation at the surface of phosphonate-functionalized polystyrene. | ||
Firstly, polymer nanoparticles with vinylphosphonic acid functionalities on their surfaces were synthesized by miniemulsion polymerization. P(S-co-VPA) nanoparticles with sizes of 140 ± 20 nm were successfully prepared (ESI, Fig. S1†). The amount of phosphorus in the copolymer was quantified by 31P-NMR spectroscopy (ESI, Fig. S2†). As expected, the content of phosphorus in the copolymers increased (from 0.9 to 9.7 mol%) with the initial concentration of vinylphosphonic acid in the miniemulsions (from 0.5 to 9.8 mol%) (Table 1). On the contrary, the apparent molecular weights decreased from 315000 to 220000 g mol−1 when vinylphosphonic acid was incorporated (ESI, Table S1†). The electron-withdrawing nature of vinylphosphonic acid reduced the electron density of the double bonds of the benzene ring, but also of the growing radical CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2. Consequently, there may be no further growth of the polymer chain, leading to low molecular weights copolymers.
CH2. Consequently, there may be no further growth of the polymer chain, leading to low molecular weights copolymers.
| Sample name | VPA in copolymera [mol%] | VPA in copolymerb [mol%] | Sample name | Ce2O3c [wt%] | Sample name | TiO2c [wt%] |
|---|---|---|---|---|---|---|
| a Theoretical content. b Determined by 31P-NMR. c Measured with TGA. | ||||||
| P0 | — | — | F0C | 4.3 | F0T | 5.4 |
| P0.01 | 0.9 | 0.5 | F0.01C | 5.7 | F1T | 8.5 |
| P0.04 | 4.8 | 4.4 | F0.04C | 7.8 | F5T | 6.5 |
| P0.10 | 9.7 | 9.8 | F0.10C | 8.9 | F10T | 9.3 |
Poly(styrene-co-vinylphosphonic acid) fibers were obtained by electrospinning of solutions of the copolymers in DMF (see corresponding SEM micrographs in ESI, Fig. S3†). The obtained fibers were randomly distributed on the clips and form a porous membrane. The porosity probably originated from the humidity in the electrospinning environment (∼50%). It is known that vapor and DMF undergo liquid–liquid phase separation and the rapid evaporation of water can leave pores on the fibers surface.33,34 The average diameter of the fibers decreased from 320 ± 60 to 270 ± 50 nm with an increasing content of vinylphosphonic acid (ESI, Table S1†).
Because the phosphonate groups should play the role of nucleating centers, it is important to quantify their amount on the surface of the fibers. Indeed, the concentration of phosphonate on the fibers surface may not reflect exactly the concentration of phosphonate in the copolymer because preferential orientation of hydrophobic groups on the surface of the fibers is possible. The elemental composition of the fiber surfaces was characterized by XPS (see atomic concentrations in ESI, Table S2†). Not surprisingly, the O 1s atomic concentration of the P(S0.96-co-VPA0.04) fibers was higher than that of the P(S0.99-co-VPA0.01) fibers, indicating the presence of higher amount of phosphonic groups. This result suggests the successful incorporation of the VPA comonomer into PS system.
The metal oxide formation occurred on the surface of the electrospun fibers by immersing into the cerium precursor solution. After a sufficient complexation time of the precursor with vinylphosphonic acid, the crystallization was induced by the addition of a precipitating agent sodium hydroxide at a controlled rate. In a previous work of our group, we had shown that metal oxide nanoparticles are directly formed on functionalized surfaces of nanoparticles, rather than formed in the solution and merely adsorbed or heterocoagulated on the polymer surface.10 We assume an analogous formation mechanism at the surface of the fibers.
The morphology and structure of the hybrid fibers obtained from aqueous solutions of Ce(III) was investigated by SEM and TEM. Fig. 2a and b show SEM micrographs at different magnifications of P(S0.90-co-VPA0.10) fibers after ceria crystallization. The presence of vinylphosphonic acid on the surface of the fibers resulted in an efficient coverage with the ceria nanoparticles, which can be correlated with the concentration of phosphonate functionalities on the fibers surface. Ceria nanoparticles formed more homogeneously on surface of the fibers with increasing VPA amount (ESI, Fig. S4†). The content of the inorganic component in the hybrid fibers, determined by thermogravimetric analysis (TGA), ranged from 4 to 9 wt% (Table 1). Oxidation of cerium in the presence of neat polystyrene fibers led to bulk crystallization consisting of large ceria particles not only on the fiber surface but also in the space between the fibers. Nucleation on the fiber surface, with almost no bulk crystallization, occurred even when using the P(S0.99-co-VPA0.01) fibers with the lowest VPA content (ESI, Fig. S5†).
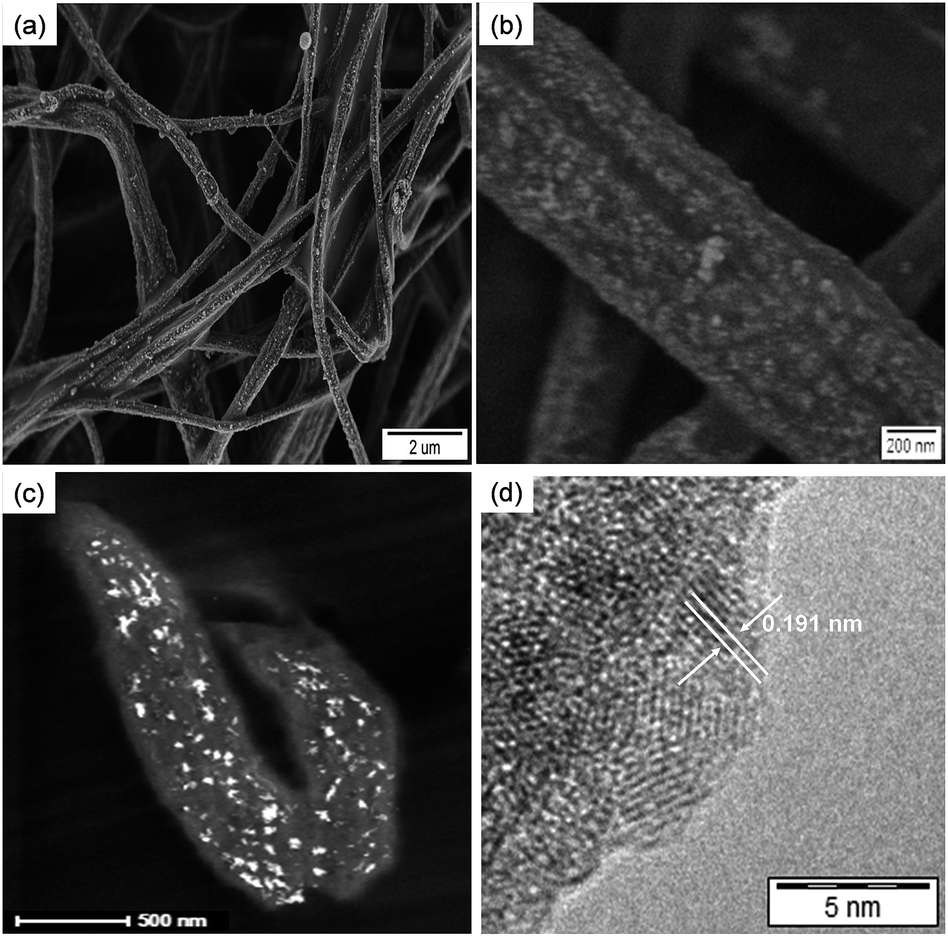 | ||
| Fig. 2 Electron micrographs of the electrospun poly(styrene-co-vinylphosphonic acid) P(S0.90-co-VPA0.10) fibers after ceria crystallization: (a and b) SEM micrographs at different magnifications, (c) TEM dark-field micrograph of a microtomed section the hybrid fibers, and (d) high-resolution TEM image. | ||
Elemental analysis by energy dispersive X-ray (EDX) spectroscopy proved the presence of cerium and oxygen in the hybrid fibers, which is attributed to the formation of ceria nanoparticles (see ESI, Fig. S6†). X-ray diffraction (XRD) patterns of the samples were dominated by a typical amorphous halo from the polymer. However, the dark-field TEM image in Fig. 2c demonstrates that ceria nanocrystals are present on the surface of the fibers. Interplanar distances of 0.191 nm measured from high-resolution TEM micrographs (Fig. 2d) correspond to the {220} planes of cubic CeO2.
Surface elemental compositions of the hybrid fibers were also investigated by XPS. The spectra, presented in Fig. 3, show characteristic signals for cerium (Ce 3d at 887.5 eV and 905 eV), oxygen (O 1s at 535.1 eV), carbon (C 1s at 286 eV), and phosphorus (P 2p 133.5 at eV). The XPS spectrum from cerium splits into Ce 3d3/2 and Ce 3d5/2 with multiple shake-up and shake-down satellites. The signals between 875 and 895 correspond to the Ce 3d5/2 while signals between 895 and 910 eV belong to the Ce 3d3/2 levels.35 The signal at 918 eV is a characteristic for the presence of cerium(IV).36 The absence of this band indicates that the oxide on the surface is mostly Ce2O3, which would be rather unusual and different from the CeO2 obtained under very similar conditions with latex particles.10 For the origin of Ce(III) oxide, two scenarios are possible: (i) since Ce2O3 is an intermediate in the formation of CeO2, its occurrence could be explained by an incomplete crystallization;37 (ii) a reduction of Ce(IV) to Ce(III), as reported by previous works.38–40 We cannot rule out any of the options, but the second appears more plausible, taking into account that the formation of CeO2 is thermodynamically favorable under our synthesis conditions.
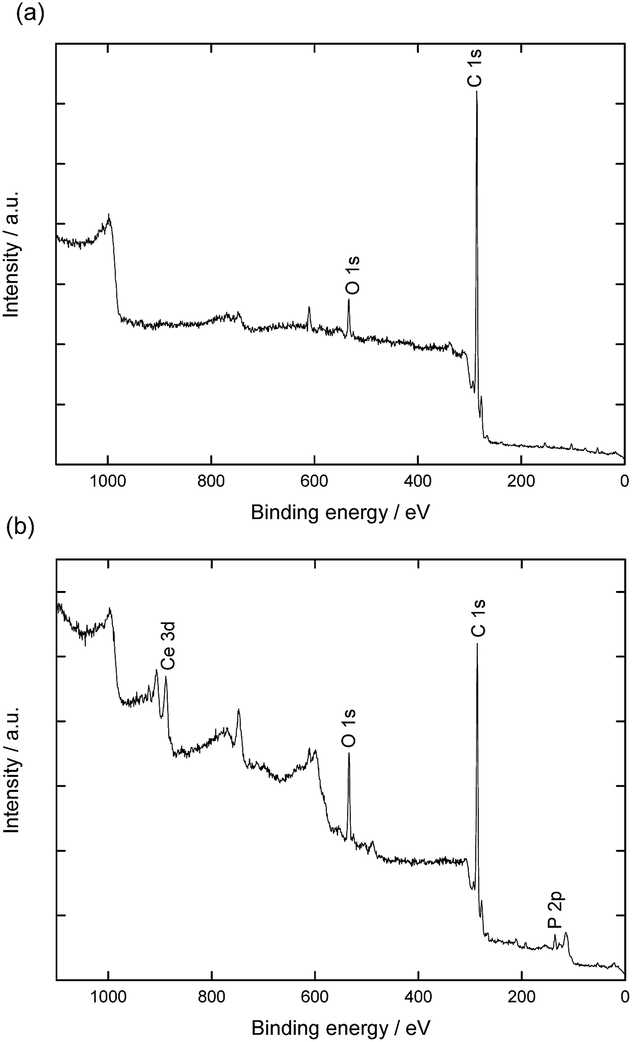 | ||
| Fig. 3 XPS spectra of the electrospun poly(styrene-co-vinylphosphonic acid) P(S0.96-co-VPA0.04) fibers (a) before and (b) after ceria crystallization. | ||
XPS measurements also indicate that phosphorus is present on the fibers on which cerium was crystallized in concentrations slightly higher than those expected for the bulk (cf. Table S2†). The concentration of oxygen atoms also increased after crystallization/washing. This result suggests reorganization of vinyl phosphonate at the molecular level during the crystallization process. Indeed, the polymer of the fibers surface experienced first the air as interface and then an aqueous medium during the crystallization.
To show the versatility of our approach, we extended the method to titania, as a representative case of a sol–gel system. The surface of the fibers was decorated with titania by adding isopropanol–titanium isopropoxide mixture to the isopropanol–water mixture containing VPA-functionalized polystyrene nanofiber. Fig. 4 shows SEM micrographs of the P(S0.99-co-VPA0.01) fibers after titania crystallization.
 | ||
| Fig. 4 SEM micrographs of the electrospun poly(styrene-co-vinylphosphonic acid) P(S0.99-co-VPA0.01) fibers after TiO2 crystallization: (a) general view, (b) higher magnification of the fiber surface. | ||
Homogeneously distributed titania nanoparticles were present on the surface of the fibers, as also supported by EDX measurements (ESI, Fig. S7†). It needs to be noted that the formation mechanisms for ceria and titania are very different: while ceria crystallizes easily at room temperature, titania follow a conventional sol–gel process and the formed material is amorphous. Therefore, the results showed that the method can be applied for the formation of hybrid fibers not only in aqueous but also in alcoholic media.
4. Conclusions
A template-assisted approach for the fabrication of polymer/metal oxide hybrid fibers is presented. Poly(styrene-co-vinylphosphonic acid) P(S-co-VPA) nanofibers were coated with ceria or titania nanoparticles at low temperature. Metal oxide nanoparticle formation could be enhanced by the introduction of functional phosphonate groups in the copolymer, which is the complexation group on the surface. Increasing the VPA concentration on the fibers surface increased the concentration of metal oxide particles on the fibers. This observation supported the role of surface VPA as nucleation centers along with fiber. The proposed synthesis strategy is simple and versatile and can be extended for the fabrication of a wide range of polymer/metal oxide hybrid membranes that are expected to provide highly reactive surfaces for enhanced catalysis, sensing, and photoelectric applications.Acknowledgements
The authors thank Kathrin Kirchhoff for TEM measurements. The financial support by the International Max Planck Research School for Polymer Materials Science (IMPRS-PMS, Mainz) is gratefully acknowledged.Notes and references
- Z. L. Wang and X. D. Feng, J. Phys. Chem. B, 2003, 107, 13563 CrossRef CAS.
- T. Tachikawa, T. Yonezawa and T. Majima, ACS Nano, 2013, 7, 263 CrossRef CAS PubMed.
- Y. Lu, M. Hoffmann, R. S. Yelamanchili, A. Terrenoire, M. Schrinner, M. Drechsler, M. W. Möller, J. Breu and M. Ballauff, Macromol. Chem. Phys., 2009, 210, 377 CrossRef CAS PubMed.
- Y. Lu, T. Lunkenbein, J. Preussner, S. Proch, J. Breu, R. Kempe and M. Ballauff, Langmuir, 2010, 26, 4176 CrossRef CAS PubMed.
- F. Polzer, E. Holub-Krappe, H. Rossner, A. Erko, H. Kirmse, F. Plamper, A. Schmalz, A. H. E. Müller and M. Ballauff, Colloid Polym. Sci., 2013, 291, 469 CAS.
- M. Agrawal, A. Pich, N. E. Zafeiropoulos, S. Gupta, J. Pionteck, F. Simon and M. Stamm, Chem. Mater., 2007, 19, 1845 CrossRef CAS.
- M. Agrawal, A. Pich, S. Gupta, N. E. Zafeiropoulos, P. Simon and M. Stamm, Langmuir, 2008, 24, 1013 CrossRef CAS PubMed.
- M. Agrawal, A. Pich, N. E. Zafeiropoulos and M. Stamm, Colloid Polym. Sci., 2008, 286, 593 CAS.
- M. Agrawal, A. Pich, S. Gupta, N. E. Zafeiropoulos, P. Formanek, D. Jehnichen and M. Stamm, Langmuir, 2010, 26, 526 CrossRef CAS PubMed.
- V. Fischer, I. Lieberwirth, G. Jakob, K. Landfester and R. Muñoz-Espí, Adv. Funct. Mater., 2013, 23, 451 CrossRef CAS PubMed.
- M. M. Demir, G. Ugur, M. A. Gulgun and Y. Z. Menceloglu, Macromol. Chem. Phys., 2008, 209, 508 CrossRef CAS PubMed.
- M. M. Demir, M. A. Gulgun, Y. Z. Menceloglu, B. Erman, S. S. Abramchuk, E. E. Makhaeva, A. R. Khokhlov, V. G. Matveeva and M. G. Sulman, Macromolecules, 2004, 37, 1787 CrossRef CAS.
- S. Madhugiri, B. Sun, P. G. Smirniotis, J. P. Ferraris and K. J. Balkus, Microporous Mesoporous Mater., 2004, 69, 77 CrossRef CAS PubMed.
- Y. W. Ju, J. H. Park, H. R. Jung, S. J. Cho and W. J. Lee, Mater. Sci. Eng., B, 2008, 147, 7 CrossRef CAS PubMed.
- Z. Y. Li, H. N. Zhang, W. Zheng, W. Wang, H. M. Huang, C. Wang, A. G. MacDiarmid and Y. Wei, J. Am. Chem. Soc., 2008, 130, 5036 CrossRef CAS PubMed.
- W. Zheng, X. F. Lu, W. Wang, Z. Y. Li, H. N. Zhang, Y. Wang, Z. J. Wang and C. Wang, Sens. Actuators, B, 2009, 142, 61 CrossRef CAS PubMed.
- S. Jeon, J. Yun, Y. S. Lee and H. I. Kim, J. Ind. Eng. Chem., 2012, 18, 487 CrossRef CAS PubMed.
- K. Lee and S. Lee, J. Appl. Polym. Sci., 2012, 124, 4038 CrossRef CAS PubMed.
- S. Sundarrajan and S. Ramakrishna, J. Mater. Sci., 2007, 42, 8400 CrossRef CAS PubMed.
- W. Yu, C. H. Lan, S. J. Wang, P. F. Fang and Y. M. Sun, Polymer, 2010, 51, 2403 CrossRef CAS PubMed.
- C. Drew, X. Liu, D. Ziegler, X. Y. Wang, F. F. Bruno, J. Whitten, L. A. Samuelson and J. Kumar, Nano Lett., 2003, 3, 143 CrossRef CAS.
- S. Nagamine, Y. Tanaka and M. Ohshima, Chem. Lett., 2009, 38, 258 CrossRef CAS.
- S. X. Shu and C. R. Li, in Materials Processing Technology, Pts 1–3, ed. X. H. Liu, Z. Jiang and J. T. Han, 2012, vol. 418–420, p. 237 Search PubMed.
- J. H. Zhang, F. H. J. Maurer and M. S. Yang, J. Phys. Chem. C, 2011, 115, 10431 CAS.
- N. Horzum, R. Muñoz-Espí, G. Glasser, M. M. Demir, K. Landfester and D. Crespy, ACS Appl. Mater. Interfaces, 2012, 4, 6338 CAS.
- S. Neubert, D. Pliszka, V. Thavasi, E. Wintermantel and S. Ramakrishna, Mater. Sci. Eng., B, 2011, 176, 640 CrossRef CAS PubMed.
- M. Roso, S. Sundarrajan, D. Pliszka, S. Ramakrishna and M. Modesti, Nanotechnology, 2008, 19, 285707 CrossRef PubMed.
- C. Drew, X. Y. Wang, F. F. Bruno, L. A. Samuelson and J. Kumar, Compos. Interfaces, 2005, 11, 711 CrossRef CAS PubMed.
- S. Nagamine, S. Ochi and M. Ohshima, Mater. Res. Bull., 2011, 46, 2328 CrossRef CAS PubMed.
- A. Ziegler, K. Landfester and A. Musyanovych, Colloid Polym. Sci., 2009, 287, 1261 CAS.
- R. Freeman, R. Kaptein and H. D. W. Hill, J. Magn. Reson., 1972, 7, 327 CAS.
- D. Crespy and K. Landfester, Beilstein J. Org. Chem., 2010, 6, 1132 CrossRef CAS PubMed.
- C.-L. Pai, M. C. Boyce and G. C. Rutledge, Macromolecules, 2009, 42, 2102 CrossRef CAS.
- M. M. Demir, eXPRESS Polym. Lett., 2010, 4, 2 CrossRef CAS.
- P. Burroughs, A. Hamnett, A. F. Orchard and G. Thornton, J. Chem. Soc., Dalton Trans., 1976, 1686 RSC.
- J. E. Castle, Surf. Interface Anal., 1984, 6, 302 CrossRef PubMed.
- O. Kepenekci, M. Emirdag-Eanes and M. M. Demir, J. Nanosci. Nanotechnol., 2011, 11, 3565 CrossRef CAS PubMed.
- E. Paparazzo, G. M. Ingo and N. Zacchetti, J. Vac. Sci. Technol., A, 1991, 9, 1416 CAS.
- M. V. R. Rao and T. Shripathi, J. Electron Spectrosc. Relat. Phenom., 1997, 87, 121 CrossRef.
- F. Zhang, P. Wang, J. Koberstein, S. Khalid and S. W. Chan, Surf. Sci., 2004, 563, 74 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM micrographs of the polymer nanoparticles, and nanofibers before and after crystallization, EDX spectrum of the nanofibers after crystallization, 31P-NMR spectra of the polymers, tables showing the diameter of the nanofibers and molecular weight of the polymer, and atomic concentrations of the nanofibers before and after ceria crystallization. See DOI: 10.1039/c5ra02140e |
| This journal is © The Royal Society of Chemistry 2015 |