DOI:
10.1039/C5RA02129D
(Paper)
RSC Adv., 2015,
5, 22818-22824
Synthesis and electrochemical performance of Na-modified Li2Fe0.5Mn0.5SiO4 cathode material for Li-ion batteries
Received
3rd February 2015
, Accepted 23rd February 2015
First published on 23rd February 2015
Abstract
A series of Li2−xNaxFe0.5Mn0.5SiO4/C (x = 0.00, 0.01, 0.03 and 0.05) composites have been synthesized via a refluxing-assisted solid-state reaction, and characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), galvanostatic charge–discharge measurements, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) tests. XRD results show that Li2−xNaxFe0.5Mn0.5SiO4/C can be well indexed as the structure of two mixed polymorphs with space group P21 and Pmn21. XPS results confirms that Na not only exists on the surface of Li2Fe0.5Mn0.5SiO4 particles, but also has been successfully doped into the crystal lattice of Li2Fe0.5Mn0.5SiO4. Na-doping can significantly improve the discharge capacity and the rate capability of Li2Fe0.5Mn0.5SiO4/C. The enhanced electrochemical performance can be attributed to the increased electronic conductivity, the decreased charge transfer impedance, and the improved Li-ion diffusion coefficient.
Introduction
Since Nytén et al. first explored Li2FeSiO4 (LFS) as a new kind of polyanion cathode material for lithium-ion batteries,1,2 LFS has attracted wide interest due to its low cost, high safety, environmentally benign, and high theoretical capacity (166 mA h g−1 for one Li+ ion exchange, and 332 mA h g−1 for two Li+ ions exchange). Furthermore, compared with LiFePO4, LFS behaves a lower band gap and a stronger Si–O bond, which is expected to get a better cycle performance.3–7 Unhappily, like other polyanion cathode materials (i.e., LiFePO4,8–11 Li3V2(PO4)3,12–14 etc.), LFS suffers from poor capability due to its poor intrinsic electronic conductivity and slow lithium ion diffusion rate, which limits its large scale application in lithium-ion batteries.3,4,6,7 Therefore, much effort has been made to improve the electrochemical performance of LFS, such as carbon coating,2–7 particle downsizing,5,6,15,16 and metal cation doping.17–21 To our knowledge, doping a small amount of metal cations can lead to lattice defects, which is beneficial to insertion/extraction of lithium ions and improving the intrinsic conductivity of cathode materials, such as LiFePO4,9,10 Li3V2(PO4)3,14,22 and so on. So far, LFS has been doped by various metal cations, such as V,17 Mn,18–20 Zn,21 Cu,21 Ni.21 Li2MnSiO4 (LMS), as an active cathode material from the Li2MSiO4 family, was first reported as a cathode material for lithium ion battery cathodes23 by Dominko et al. In the past few years, LMS has drawn wide attention due to its high theoretical capacity (334 mA h g−1) and high operating voltage (>4.2 V).24,25 Unfortunately, LMS is unstable upon delithiation with a strong tendency to be amorphous. It has been proved that Li2FexMn1−xSiO4 with Fe/Mn = 1 can obtain a desirable electrochemical performance.20,27,28 For instance, Z. L. Gong et al.27 first reported a high capacity of 214 mA h g−1 can be achieved for Li2MnxFe1−xSiO4 at x = 0.5. LFS is isostructural with LMS (orthorhombic with a space group of Pmn21), so it is easy to form Li2MnxFe1−xSiO4 solid solutions.27 C. Deng and B. Shao et al. studied the electrochemical performance of Li2Fe1−xMnxSiO4/C (x = 0, 0.3, 0.5, 0.7, 1) and Li2FexMn1−xSiO4/C (0≦x≦0.8), and also found that the Li2Fe0.5Mn0.5SiO4/C sample exhibited the maximum discharge capacity.20,28 However, the cycling performance of the Li2FexMn1−xSiO4/C samples decreases with decreasing x due to the low electronic conductivity and the low stability of LMS.28 However, Li2Mn0.5Fe0.5SiO4 also requires further modifications to overcome limitations such as slow lithium-ion diffusion and low electronic conductivity. To best of our knowledge, cation doping is an efficient way to improve the intrinsic electronic conductivity and chemical diffusion coefficient of lithium ions within the crystals. As for Na-doping, it is confirmed that an appropriate amount of Na-doping at Li-site can effectively improve the electrochemical performance of LiFePO4, Li3V2(PO4)3 and LiNi1/3Co1/3Mn1/3O2.29–32 But, there is no report about Na-doping for Li2Fe0.5Mn0.5SiO4 cathode material.
In this work, we first designed Na-doping at Li-site for Li2Fe0.5Mn0.5SiO4, therefore a series of Na-doped Li2Fe0.5Mn0.5SiO4 composites were prepared via a refluxing-assisted solid-state reaction, and characterized with X-ray diffraction (XRD), scanning electron microscopy (SEM), and X-ray photoelectron spectroscopy (XPS). The effect of Na-incorporation on the electrochemical performance of Li2Fe0.5Mn0.5SiO4/C was also investigated by galvanostatic charge–discharge measurements, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) tests.
Experimental
Li2−xNaxFe0.5Mn0.5SiO4/C (x = 0.00, 0.01, 0.03 and 0.05) composites were synthesized via a refluxing-assisted solid-state reaction. All chemicals were of analytical grade and used without further purification. A stoichiometric amount of tetraethyl orthosilicate (TEOS), CH3COOLi·2H2O, FeC2O4·2H2O, C4H6MnO4·2H2O, and NaNO3 were dispersed in ethanol. The above mixture was refluxed at 80 °C for 24 h under stirring till a brown gel was formed. The resulting wet gel was dried at 50 °C over night. The obtained dry gel was finely ground with 10 wt% sucrose in acetone for 7 h. After drying, the above mixture was calcined at 350 °C for 5 h, and then sintered at 650 °C for 10 h under flowing nitrogen gas. After natural cooling down to room temperature, the powders were ground and sieved to obtain the final products. The Li2−xNaxFe0.5Mn0.5SiO4/C composites with x = 0.00, 0.01, 0.03 and 0.05 will be referred as LFMS, LFMS-0.01Na, LFMS-0.03Na, LFMS-0.05Na, respectively.
The phase identification of the obtained samples was performed by powder X-ray diffraction (XRD, Rigaku Ultima IV) employing Cu-kα radiation (λ = 1.5406 Å). Diffraction patterns were scanned over the range of 2θ between 10° and 80°. The morphology was observed with a field-emission scanning electron microscope (FESEM, JSM-7500F, JEOL). The oxidation state of key elements (i.e., Fe, Mn and Na) in LFMS-0.01Na was studied by X-ray photoelectron spectroscopy (XPS, PHI Quantera, U-P). In order to investigate the distribution of key elements (C, Si, Fe, Mn and Na) in LFMS-0.01Na, Ar-ion sputtering was also used in XPS measurement. Electrical conductivity was measured with a standard four-probe method by RTS resistivity measurement system (RTS-8, China) on disk-shaped pellets with diameter of 8 mm and thickness of about 1.0 mm. The amount of residual carbon was tested by an IR carbon/sulfur determinator with high frequency induction combustion furnace (HW2000B).
The electrochemical properties of the obtained samples were measured in CR2025 coin cells using lithium foil as counter and reference electrodes. The coin cells were prepared as described in ref. 7. The working electrodes were prepared by mixing active materials (75 wt%), acetylene black (15 wt%) and polyvinylidene fluoride (PVDF, 10 wt%) in N-methyl pyrrolidinone (0.02 g mL−1) on an aluminum foil (20 μm in thickness) which was used as the current collectors. The loading of the active materials on the electrode was 1.8 mg cm−2. Galvanostatic charge–discharge measurements were performed in a voltage range of 1.5–4.6 V on a battery test system (LAND CT2001A, China). All reported capacities are quoted with respect to the mass of the obtained samples including the coating carbon. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurement was performed on an electrochemical working station (CHI614C, China) over a frequency range between 0.01 Hz and 100 kHz.
Results and discussion
Fig. 1 shows the XRD patterns of LFMS and LFMS-0.01Na samples. A full Rietveld refinement was carried out by software Maud. The best refinement models were chosen from P21 space group (LFS) and Pmn21 space group (LMS). It can be clearly seen that both samples show a similar XRD pattern. Though some impurities such as Fe2SiO4 (∼30°)26 and Mn2SiO4 (∼35°)24,25 appear in the XRD patterns of LFMS and LFMS-0.01Na, the main diffraction peaks of both samples are well indexed as the structure of two mixed polymorphs with space group P21 and Pmn21,18 which indicates that Na-incorporation has no inherent effect on the Li2Fe0.5Mn0.5SiO4 phase formation. The refined lattice parameters and atomic coordination are listed in Tables 1 and 2, respectively. Because the reliability factor of sig is less than 2, and Rw is less than 15% (For LFMS, the reliable factors are Rw = 12.15%, sig = 1.86; and for LFMS-0.01Na, the reliable factors are Rw = 11.88%, sig = 1.82), the Rietveld refinement results are reliable in the following analysis of crystal structure. As shown in Table 1, after Na-incorporation, the cell volume of LMS slightly decreases, whereas that of LFS increases. As we know, the cell volume changes with the Na-doping cannot be fully explained by the atom size difference.33 Furthermore, no peaks for crystalline carbon are observed in both XRD patterns, suggesting that the residual carbon in LFMS and LFMS-0.01Na composites is in amorphous form. The amount of residual carbon in Li2−xNaxFe0.5Mn0.5SiO4/C with x = 0.00, 0.01, 0.03 and 0.05 is about 8.5, 8.1, 8.0, and 8.4 wt%, respectively.
 |
| | Fig. 1 XRD patterns of (a) LFMS, and (b) LFMS-0.01Na. | |
Table 1 Lattice parameters of LFMS and LFMS-0.01Na
| Sample |
Phase |
a (Å) |
b (Å) |
c (Å) |
V (Å3) |
| LFMS |
LFS |
8.2701 |
4.9813 |
8.2770 |
340.98 |
| LMS |
6.2813 |
5.3864 |
5.0070 |
169.40 |
| LFMS-0.01Na |
LFS |
8.2683 |
5.0127 |
8.2799 |
343.17 |
| LMS |
6.2732 |
5.3915 |
4.9929 |
168.87 |
Table 2 Atomic fractional coordinates of LFMS and LFMS-0.01Na
| Phase |
Atom |
LFMS |
LFMS-0.01Na |
| X |
Y |
Z |
X |
Y |
Z |
| LMS |
Li1 |
0.7562 |
−0.1542 |
1.1387 |
0.7566 |
−0.1401 |
1.1206 |
| Mn1 |
0.5000 |
0.3677 |
0.9998 |
0.5000 |
0.3599 |
1.0115 |
| Si1 |
1.0000 |
0.3193 |
1.0178 |
1.0000 |
0.3239 |
1.0195 |
| O1 |
0.5000 |
0.6765 |
1.1607 |
0.5000 |
0.6651 |
1.1416 |
| O2 |
0.5000 |
0.3257 |
0.5521 |
0.5000 |
0.3274 |
0.5585 |
| O3 |
0.7614 |
0.1641 |
1.0829 |
0.7651 |
0.1684 |
1.0928 |
| LFS |
Li1 |
0.6570 |
0.7763 |
0.6750 |
0.6166 |
0.6857 |
0.6526 |
| Li2 |
0.6012 |
0.0006 |
0.0752 |
0.6057 |
−0.0870 |
0.1167 |
| Fe1 |
0.2785 |
0.7905 |
0.5467 |
0.2660 |
0.8196 |
0.5283 |
| Si1 |
0.0249 |
0.7843 |
0.7670 |
0.0261 |
0.8151 |
0.7725 |
| O1 |
0.8549 |
0.6593 |
0.8572 |
0.8580 |
0.6967 |
0.8484 |
| O2 |
0.4081 |
0.2263 |
0.8687 |
0.3981 |
0.2436 |
0.8245 |
| O3 |
0.6831 |
0.7596 |
0.5011 |
0.6678 |
0.7751 |
0.4848 |
| O4 |
0.9587 |
0.8002 |
0.2337 |
0.9444 |
0.8000 |
0.2360 |
Fig. 2 shows the SEM images of Li2−xNaxFe0.5Mn0.5SiO4/C samples. As shown in Fig. 2, there is no significant difference in the morphology between the four samples. All the samples present irregular granular shape with a receivable size distribution ranging from ∼100 nm to ∼500 nm.
 |
| | Fig. 2 SEM images of (a) LFMS, (b) LFMS-0.01Na, (c) LFMS-0.03Na, and (d) LFMS-0.05Na. | |
X-ray photoelectron spectroscopy (XPS) is a useful tool to study the oxidation state of key elements in samples, and also an important surface analysis technique to investigate the element distribution.14 Fig. 3a shows the typical survey XPS spectrum of LFMS-0.01Na, and Fig. 3b–f show the high-resolution spectra of C1s, Si2p, Fe2p3, Mn2p3 and Na1s, respectively. The obtained binding energy (BE) in the XPS analysis was referenced by setting the BE of C1s to 284.5 eV. The intensity of C1s on the surface is much stronger than that in the interior (Fig. 3b), which reveals that carbon is mainly coated on the surface of the LFMS particles. Instead, the intensity of Si2p, Fe2p3 and Mn2p3 main peaks (Fig. 3c–e) on the surface is much lower than that in the interior due to the carbon coating layer. From Fig. 2f, it is worthwhile to note that Na1s main peak appears not only on the surface but also is clearly observed in the interior. Therefore, it is reasonable to believe that some Na exists on the surface in a form of Na-containing composite (i.e., Na4SiO4) though no Na-containing composite is detected in the XRD pattern (Fig. 1) because of the low-level amount; another part of Na should enter into the lattice of LFMS. Furthermore, the Fe2p3 main peak at ∼711 eV is very close to that for the Fe2+ in LiFePO4,34,35 which indicate that Na-incorporation don't change the divalent state of Fe in LFMS. The Mn2p3 main peak at ∼641 eV is consistent with that of Mn2+ in LMS,24 confirming that the oxidation state of Mn in LFMS-0.01Na is +2. Noting that, Fe peaks at surface are different from that in interior. Due to the chemical reduction of Ar-ion sputtering,36,37 the XPS peak (706.5 eV) in the interior is related to elemental Fe.38 To our knowledge, the electrode potential of Fe2+/Fe is more positive than that of Mn2+/Mn, that is to say, Fe2+ can be reduced more easily than Mn2+, thus no elemental Mn peak appears in the internal XPS spectra (Fig. 3e). In addition, the binding energy of Na1s (∼1071 eV) for LFMS-0.01Na is very close to that for Na+ in Na2HPO4,38 which indicates that the oxidation state of Na in LFMS-0.01Na is +1.
 |
| | Fig. 3 XPS spectra of LFMS-0.01Na. | |
Galvanostatic charge–discharge measurements were carried out at room temperature to investigate the effect of Na-incorporation on the electrochemical performance of LFMS, LFMS-0.01Na, LFMS-0.03Na and LFMS-0.05Na. Fig. 4 shows the first two charge–discharge profiles at 0.1 C (1 C = 166 mA h g−1) in the voltage range of 1.5–4.6 V (vs. Li+/Li). The second charge plateau is obvious lower than the first one, which suggested that a structural rearrangement might occur during the initial charge process.2 As shown in Fig. 4, the LMFS-0.01Na electrode delivers the highest initial specific capacity of 264.6 mA h g−1, corresponding to 1.59 mol of Li+-ion per formula unit. Obviously, LMFS-0.01Na exhibits higher initial specific capacity than other three samples (175.5 mA h g−1 for LMFS, 187.9 mA h g−1 for LMFS-0.03Na, and 173.1 mA h g−1 for LMFS-0.05Na).
 |
| | Fig. 4 Charge–discharge profiles of the as-prepared samples. | |
Fig. 5 shows the cycle performance of LFMS, LFMS-0.01Na, LFMS-0.03Na and LFMS-0.05Na electrodes at 0.1 C. As seen in Fig. 5, the discharge capacity of all the four cathodes gradually decreased due to the increased polarization. It is found that all the Na-doped LFMS composites show enhanced discharge capacity, i.e., after 20 cycles, LFMS-0.01Na delivers the highest capacity of 136.9 mA h g−1; and LFMS-0.03Na and LFMS-0.05Na show moderate capacities of 110.3 mA h g−1 and 95.5 mA h g−1, respectively; whereas LFMS only exhibits the lowest capacity of 84.0 mA h g−1. The increased capacity by Na-doping is related to the pillar effect of sodium ions, which can provide larger space for the movement of lithium ions.29 Considering the structural rearrangement during the initial charge process, we chose the discharge capacity of the second cycle to calculate the capacity retention. The capacity retention ratio of LFMS is calculated to be 47.9%, whereas the capacity retention ratio of LFMS-0.01Na, LFMS-0.03Na and LFMS-0.05Na is slightly increased to 51.7, 58.7 and 55.1%, respectively. Obviously, after Na-incorporating, the discharge capacity of LFMS is significantly enhanced, but the capacity retention ratio is only slightly improved. The large capacity fade can be attributed to the amorphous transition of LMS in Li2−xNaxFe0.5Mn0.5SiO4/C (x = 0.01, 0.03 and 0.05), in that the amorphous transition of LMS might have made the lithium ion diffusion routes in crystal LMS particles disordered, thus making it difficult for lithium ions to insert inside the LMS particles.28 To proof this point, the XRD patterns of LFMS before and after charging/discharging were shown in Fig. 6. Obviously, after charging/discharging, the diffraction peaks become weaker and even disappear, which demonstrate that LMS in LFMS changed to an amorphous state on the first charge,27 and the amorphous transition process of LMS is irreversible, thus a lower capacity retention ratio.
 |
| | Fig. 5 Cycle performance curves of the as-prepared samples at 0.1 C. | |
 |
| | Fig. 6 XRD patterns of the LFMS before charge, charge to 4.6 V and discharge to 1.5 V, respectively. | |
Fig. 7 shows the rate performance of LFMS, LFMS-0.01Na, LFMS-0.03Na and LFMS-0.05Na electrodes, which were tested in a mode such that all cells were charged under a small current density of 0.1 C to 4.6 V and discharged at different rates (0.5 C, 1 C, 2 C, 5 C and 0.5 C) to 1.5 V, and the cell ran for 10 cycles at each current density. At 0.5 C, 1 C and 2 C, the three Na-modified LFMS electrodes all show higher capacity than un-modified LFMS. It can be explained that Na+ ions can act as pillars in the Li2−xNaxFe0.5Mn0.5SiO4/C (x = 0.01, 0.03 and 0.05) structures, which can provide larger space for the movement of lithium ions and enhance the electronic conductive property and the ionic transport feature, thus leading to an increased Li+-ion diffusion coefficient and an improved rate performance.29 When the charge–discharge rate increased to 5 C, the electrochemical performance of Li2−xNaxFe0.5Mn0.5SiO4/C samples has close connection with the electronic conductivity,29 so all the four Li2−xNaxFe0.5Mn0.5SiO4/C samples show nearly equal capacity because of the similar electronic conductivity (Table 3). When back to 0.5 C, all the Na-modified LFMS samples restore higher capacity than LFMS, because the electrochemical performance is mainly controlled by Li+-ion diffusion at low C-rate.
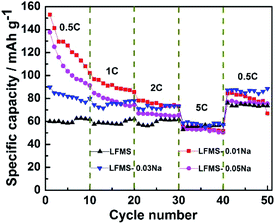 |
| | Fig. 7 Rate performance of the as-prepared samples. | |
Table 3 The electronic conductivity of samples
| Sample |
Electronic conductivity (S cm−1) |
| LFMS |
2.02 × 10−3 |
| LFMS-0.01Na |
4.59 × 10−3 |
| LFMS-0.03Na |
7.19 × 10−3 |
| LFMS-0.05Na |
6.80 × 10−3 |
The electrochemical impedance spectroscopies (EIS) for LFMS, LFMS-0.01Na, LFMS-0.03Na and LFMS-0.05Na composites are shown in Fig. 8. All EIS spectra consist of a small intercept, a depressed semicircle and an inclined line. The small intercept at the Z′ axis in the high frequency region corresponds to the ohmic resistance, representing the resistance of the electrolyte. The depressed semicircle in the medium frequency region is related to the charge transfer resistance and the double-layer capacitance between the electrolyte and cathode. The inclined line in the low frequency region is the Warburg impedance, which is associated with Li-ion diffusion in the cathode active particles.7 All EIS curves were fitted by an equivalent circuit composed of “R(C(Rw))” using the ZSimpWin program,14 and the fitting results were shown Table 4. The smaller the diameter, the lower the charge-transfer resistance is. From Fig. 8a and Table 4, it is found that the charge transfer resistance decreases after Na-incorporating, and LFMS-0.01Na (Rct = 26.12 Ω) shows the lowest charge-transfer resistance than LFMS and other Na-modified LFMS samples (i.e., 47.85 Ω for LFMS, 28.19 Ω for LFMS-0.03Na, and 33.40 Ω for LFMS-0.05Na). The effect of Na-incorporation on the charge-transfer resistance is similar to the effect on the measured electronic conductivity (4.59 × 10−3 S cm−1 for LFMS-0.01Na, but 2.02 × 10−3 S cm−1 for LFMS) (shown in Table 3). The exchange current density (i) and the diffusion coefficient of lithium ions (DLi) can be obtained according to the following equations:4,7
| | |
DLi = R2T2/2A2n4F4CLi2δ2
| (2) |
where
R is the gas constant,
T is the absolute temperature,
A is the surface area of the cathode,
n is the number of electrons per molecule during oxidation,
F is the Faraday constant,
CLi is the concentration of lithium ion.
δ is the Warburg coefficient which is related to
Z′:
4,7| | |
Z′ = RC + Rct + δω−1/2
| (3) |
where
ω is the angular frequency in the low frequency region, both
RC and
Rct are kinetics parameters independent of frequency, so
δ is also the slope for the plot of
Z′
vs. the reciprocal square root of the lower angular frequencies (
ω−1/2). To obtain the Warburg coefficient (
δ), the linear fitting of
Z′
vs. ω−1/2 in the low frequency region of all the as-prepared samples is shown in
Fig. 8b. As listed in
Table 4, LFMS-0.01Na shows the highest exchange current density (
i = 0.984 mA cm
−2) and diffusion coefficient of lithium ions (
DLi = 9.8 × 10
−12 cm
2 s
−1) compared to LFMS and other Na-modified samples (For LFMS,
i = 0.537 mA cm
−2 and
DLi = 4.5 × 10
−12 cm
2 s
−1; for LFMS-0.03Na,
i = 0.912 mA cm
−2 and
DLi = 8.5 × 10
−12 cm
2 s
−1, and for LFMS-0.05Na,
i = 0.769 mA cm
−2 and
DLi = 7.2 × 10
−12 cm
2 s
−1). Higher exchange current density and increased diffusion coefficient of lithium ions indicates faster kinetics of the cell reactions in Na-modified LFMS electrodes, which agrees well with the results of electrochemical performance tests.
 |
| | Fig. 8 (a) EIS curves, and (b) relationship between Z′ and ω−1/2 in the low frequency region of the as-prepared samples. | |
Table 4 EIS parameters of the as-prepared samples
| Sample |
Rct (Ω) |
δ (Ω s1/2) |
i (mA cm−2) |
DLi (cm2 s−1) |
| LFMS |
47.85 |
57.49 |
0.537 |
4.5 × 10−12 |
| LFMS/C-0.01Na |
26.12 |
39.09 |
0.984 |
9.8 × 10−12 |
| LFMS/C-0.03Na |
28.19 |
41.90 |
0.912 |
8.5 × 10−12 |
| LFMS/C-0.05Na |
33.40 |
45.34 |
0.769 |
7.2 × 10−12 |
To understand the effect of Na-incorporation on the electrochemical behavior of LFMS, cyclic voltammogram (CV) tests were also carried out. Fig. 9 shows the CV curves of the four as-prepared samples. Obviously, the Na-doped LFMS electrodes display the same shapes of CV curves with LFMS electrode, demonstrating that Na-incorporation does not change the electrochemical behavior of LFMS. Noting that, an extra cathodic peak at ∼1.8 V is also observed, which should be ascribed to the reaction of forming the solid electrolyte interface (SEI) film on the positive electrode surface or to some extra side reaction.39
 |
| | Fig. 9 CV profiles of the as-prepared samples. | |
Conclusions
Li2−xNaxFe0.5Mn0.5SiO4/C composites were successfully synthesized via a refluxing-assisted solid-state reaction, and its electrochemical performance was also investigated. The XRD results show that Na-incorporation has no inherent effect on the LFMS phase formation, and the main diffraction peaks of samples are well indexed as the structure of two mixed polymorphs with space group P21 and Pmn21. XPS confirms that Na not only exists on the surface of LFMS particles, but also has been successfully doped into the crystal lattice of LFMS. EIS results demonstrate that Na-modified LFMS show higher exchange current density and increased diffusion coefficient of lithium ions than the pristine LFMS. As a result, Na-modified LFMS deliver higher discharge capacity than LFMS. This work demonstrates that an appropriate Na-incorporation is an efficient way for LFMS to improve discharge capacity and rate capability.
Acknowledgements
This work was supported by the National Science Foundation of China (51302153, 51272128); the Key Project of Hubei Provincial Department of Education (D20131303); the Opening Project of CAS Key Laboratory of Materials for Energy Conversion (CKEM131404); the Scientific Fund of China Three Gorges University (KJ2012B043); the Research Innovation Foundation of Master Dissertation of China Three Gorges University (2013CX028); and the Faculty Research Grant (FRG) from Sam Houston State University.
Notes and references
- A. Nytén, A. Abouimrane, M. Armand, T. Gustafsson and J. O. Thomas, Electrochem. Commun., 2005, 7, 156 CrossRef PubMed.
- A. Nytén, S. Kamali, L. Häggström, T. Gustafsson and J. O. Thomas, J. Mater. Chem., 2006, 16, 2266 RSC.
- H. Zhu, X. Wu, L. Zan and Y. Zhang, Electrochim. Acta, 2014, 117, 34 CrossRef CAS PubMed.
- G. Peng, L. L. Zhang, X. L. Yang, S. Duan, G. Liang and Y. H. Huang, J. Alloys Compd., 2013, 570, 1 CrossRef CAS PubMed.
- Z. L. Gong, Y. X. Li, G. N. He, J. Li and Y. Yang, Electrochem. Solid-State Lett., 2008, 11, A60 CrossRef CAS PubMed.
- H. Zhou, M. A. Einarsrud and F. Vullum-Bruer, J. Power Sources, 2013, 235, 234 CrossRef CAS PubMed.
- L. L. Zhang, S. Duan, X. L. Yang, G. Peng, G. Liang, Y. H. Huang, Y. Jiang, S. B. Ni and M. Li, ACS Appl. Mater. Interfaces, 2013, 5, 12304 CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS PubMed.
- L. X. Yuan, Z. H. Wang, W. X. Zhang, X. L. Hu, J. T. Chen, Y. H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269 CAS.
- S. Y. Chung and Y. M. Chiang, Electrochem. Solid-State Lett., 2003, 6, A278 CrossRef CAS PubMed.
- G. Qin, Q. Ma and C. Wang, Electrochim. Acta, 2014, 115, 407 CrossRef CAS PubMed.
- H. Huang, S. C. Yin, T. Kerr, N. Taylor and L. F. Nazar, Adv. Mater., 2002, 14, 1525 CrossRef CAS.
- Y. Q. Qiao, J. P. Tu, X. L. Wang, D. Zhang, J. Y. Xiang, Y. J. Mai and C. D. Gu, J. Power Sources, 2011, 196, 7715 CrossRef CAS PubMed.
- L. L. Zhang, G. Liang, G. Peng, Y. H. Huang, L. Wang, L. Qie, M. C. Croft, A. Ignatov and J. B. Goodenough, J. Electrochem. Soc., 2012, 159, A1573 CrossRef CAS PubMed.
- Z. Zheng, Y. Wang, A. Zhang, T. Zhang, F. Cheng, Z. Tao and J. Chen, J. Power Sources, 2012, 198, 229 CrossRef CAS PubMed.
- J. Cui, C. Qing, Q. Zhang, C. Su, X. Wang, B. Yang and X. Huang, Ionics, 2014, 20, 23 CrossRef CAS.
- H. Hao, J. Wang, J. Liu, T. Huang and A. Yu, J. Power Sources, 2012, 210, 397 CrossRef CAS PubMed.
- R. Chen, R. Heinzmann, S. Mangold, V. S. K. Chakravadhanula, H. Hahn and S. Indris, J. Phys. Chem. C, 2013, 117, 884 CAS.
- H. Guo, X. Cao, X. Li, L. Li, X. Li, Z. Wang, W. Peng and Q. Li, Electrochim. Acta, 2010, 55, 8036 CrossRef CAS PubMed.
- C. Deng, S. Zhang and S. Y. Yang, J. Alloys Compd., 2009, 487, L18 CrossRef CAS PubMed.
- C. Deng, S. Zhang, S. Y. Yang, B. L. Fu and L. Ma, J. Power Sources, 2011, 196, 386 CrossRef CAS PubMed.
- Y. Z. Dong, Y. M. Zhao and H. Duan, J. Electroanal. Chem., 2011, 660, 14 CrossRef CAS PubMed.
- R. Dominko, M. Bele, M. Gaberšček, A. Meden, M. Remškar and J. Jamnik, Electrochem. Commun., 2006, 8, 217 CrossRef CAS PubMed.
- S. Devaraja, M. Kuezmaa, C. T. Ng and P. Balaya, Electrochim. Acta, 2013, 102, 290 CrossRef PubMed.
- V. Aravindan, S. Ravi, W. S. Kim, S. Y. Lee and Y. S. Lee, J. Colloid Interface Sci., 2011, 355, 472 CrossRef CAS PubMed.
- X. Jiang, H. Xu, J. Yang, J. Liu, H. Mao and Y. Qian, RSC Adv., 2014, 4, 39889 RSC.
- Z. L. Gong, Y. X. Li and Y. Yang, Electrochem. Solid-State Lett., 2006, 9, A542 CrossRef CAS PubMed.
- B. Shao, Y. Abe and I. Taniguchi, Powder Technol., 2013, 235, 1 CrossRef CAS PubMed.
- Q. Kuang, Y. Zhao and Z. Liang, J. Power Sources, 2011, 196, 10169 CrossRef CAS PubMed.
- C. Gong, W. Lv, L. Qu, O. E. Bankole, G. Li, R. Zhang, M. Hu and L. Lei, J. Power Sources, 2014, 247, 151 CrossRef CAS PubMed.
- X. Yin, K. Huang, S. Liu, H. Wang and H. Wang, J. Power Sources, 2010, 195, 4308 CrossRef CAS PubMed.
- W. Wang, Z. Chen, J. Zhang, C. Dai, J. Li and D. Ji, Electrochim. Acta, 2013, 103, 259 CrossRef CAS PubMed.
- L. L. Zhang, S. Duan, X. L. Yang, G. Liang, Y. H. Huang, X. Z. Cao, J. Yang, S. B. Ni and M. Li, Sci. Rep., 2014, 4, 5064 Search PubMed.
- C. S. Sun, Z. Zhou, Z. G. Xu, D. G. Wang, J. P. Wei, X. K. Bian and J. Yan, J. Power Sources, 2009, 193, 841 CrossRef CAS PubMed.
- R. Dedryvère, M. Maccario, L. Croguennec, F. L. Cras, C. Delmas and D. Gonbeau, Chem. Mater., 2008, 20, 7164 CrossRef.
- S. Hashimoto, J. Surf. Anal., 2003, 10, 230 CAS.
- T. Choudhury, S. O. Saied, J. L. Sullivan and A. M. Abbot, J. Phys. D: Appl. Phys., 1989, 22, 1185 CrossRef CAS.
- C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corporation Physical Electronics Division, Minnesota, 1979 Search PubMed.
- L. L. Zhang, H. B. Sun, X. L. Yang, Y. W. Wen, Y. H. Huang, M. Li, G. Peng, H. C. Tao, S. B. Ni and G. Liang, Electrochim. Acta, 2015, 152, 496 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.